

Combined presentation and immunogenicity analysis reveals a recurrent RAS.Q61K neoantigen in melanoma
- PMID: 34651586
- PMCID: PMC8516471
- DOI: 10.1172/JCI129466
Combined presentation and immunogenicity analysis reveals a recurrent RAS.Q61K neoantigen in melanoma
Abstract
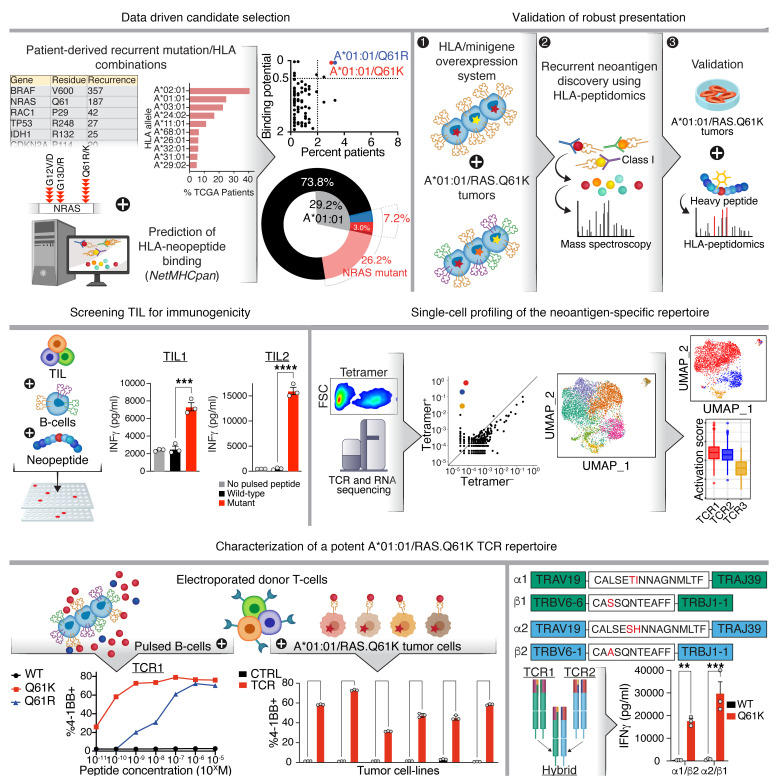
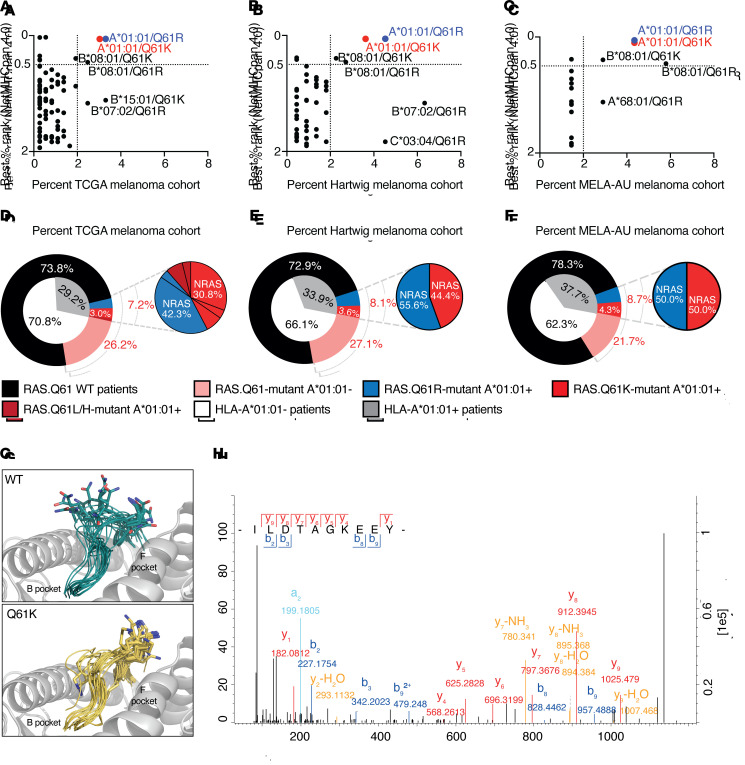
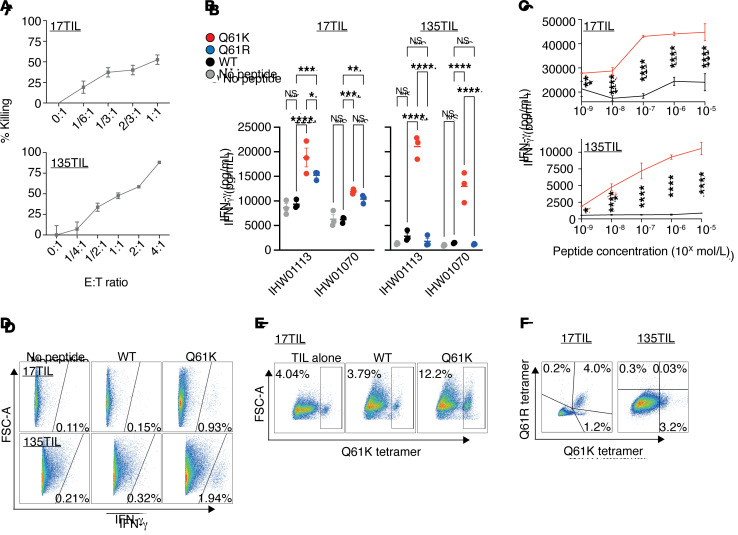
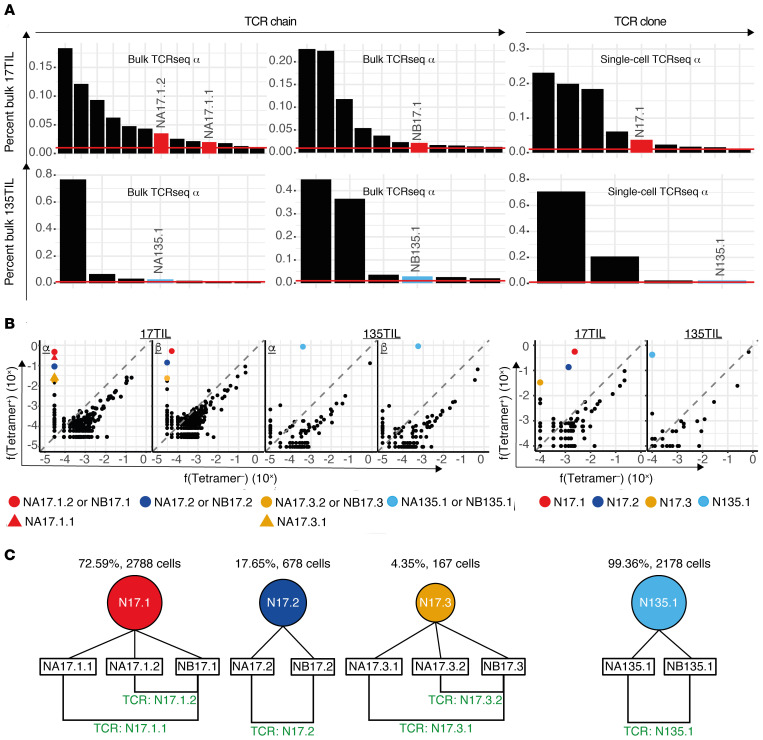
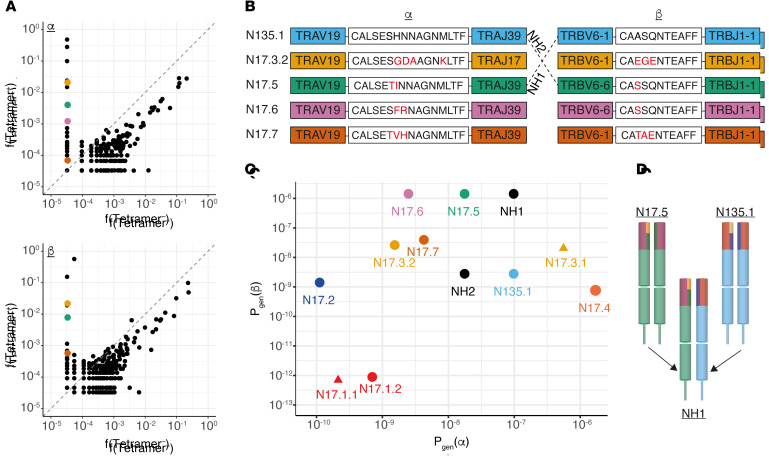
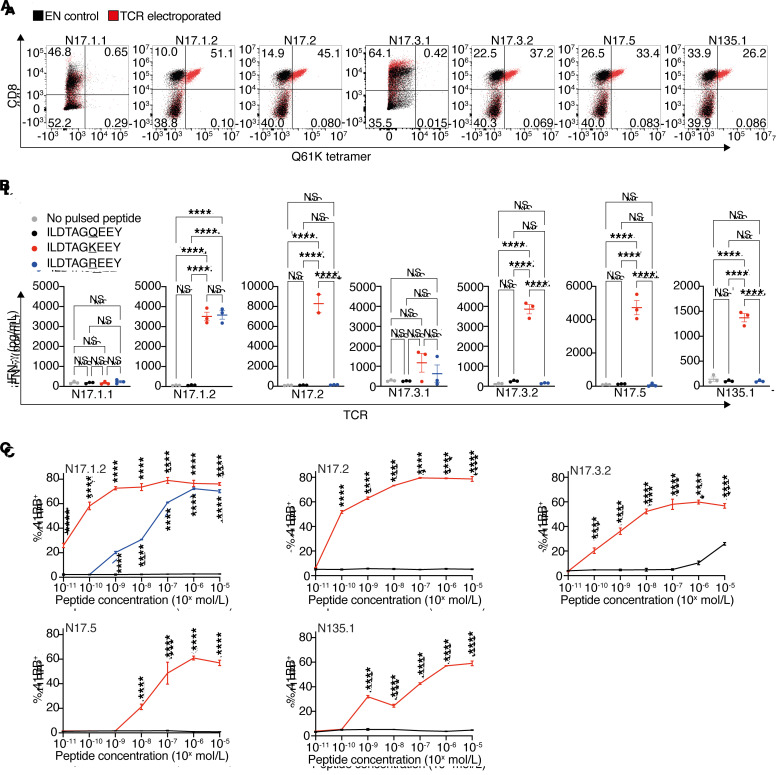
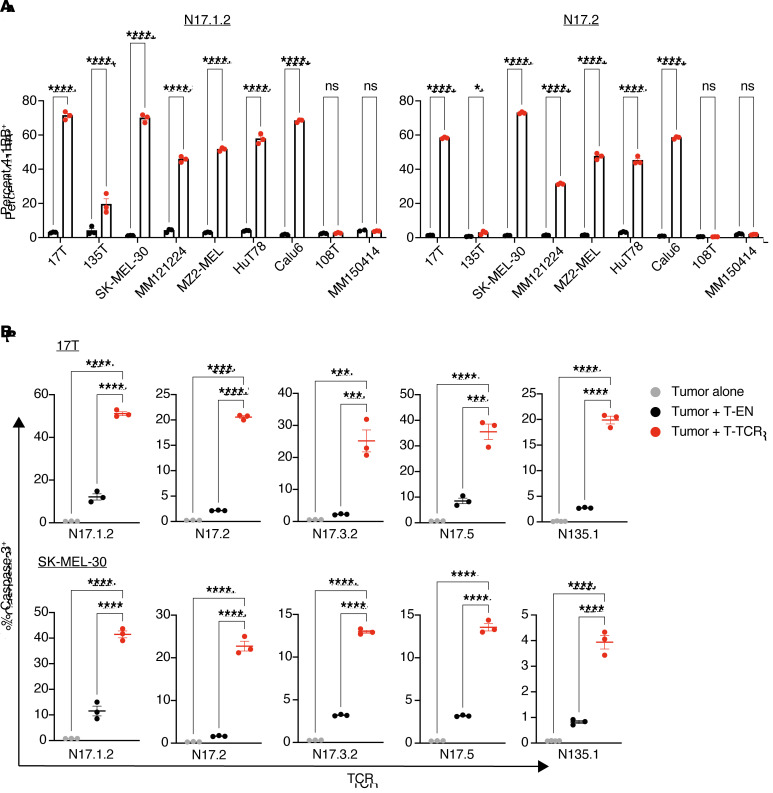
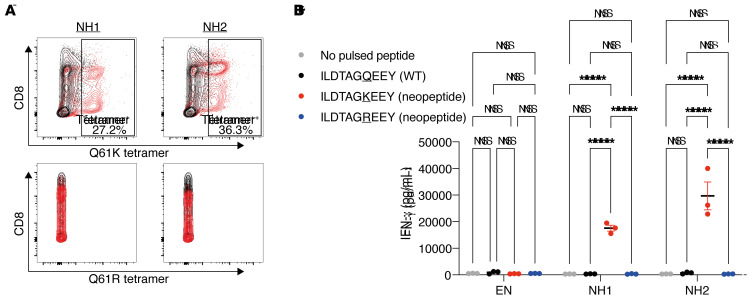
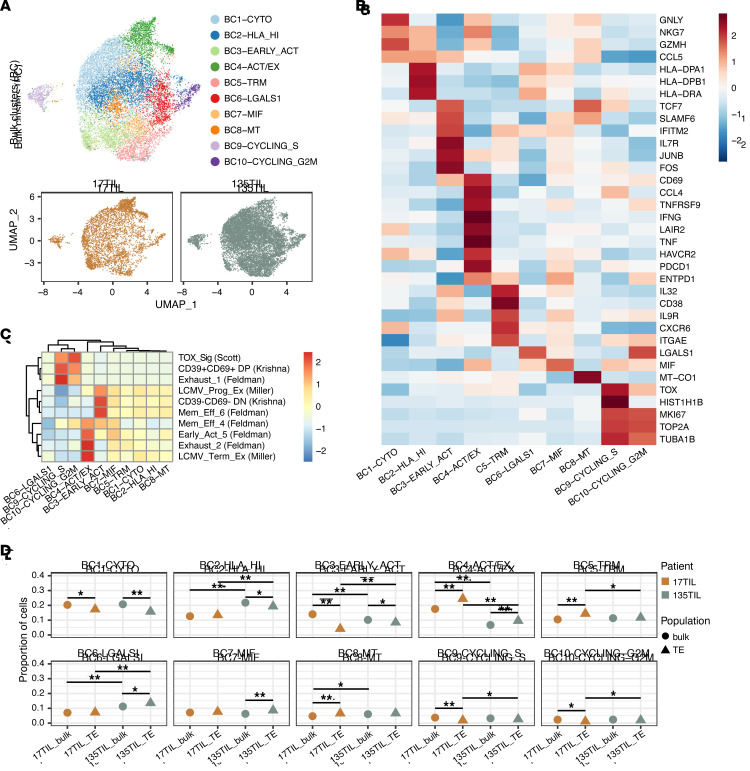
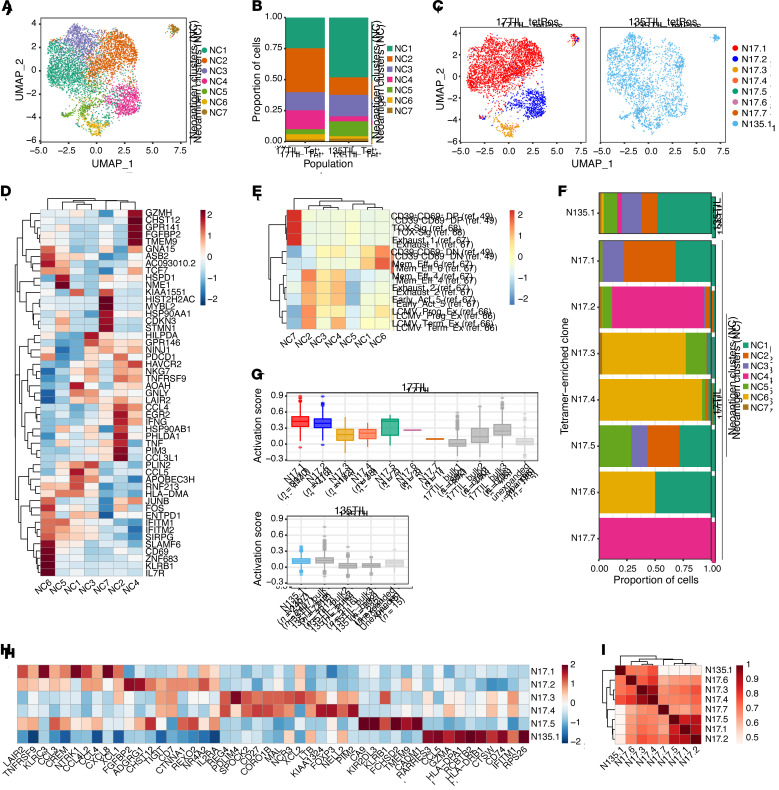
Neoantigens are now recognized drivers of the antitumor immune response. Recurrent neoantigens, shared among groups of patients, have thus become increasingly coveted therapeutic targets. Here, we report on the data-driven identification of a robustly presented, immunogenic neoantigen that is derived from the combination of HLA-A*01:01 and RAS.Q61K. Analysis of large patient cohorts indicated that this combination applies to 3% of patients with melanoma. Using HLA peptidomics, we were able to demonstrate robust endogenous presentation of the neoantigen in 10 tumor samples. We detected specific reactivity to the mutated peptide within tumor-infiltrating lymphocytes (TILs) from 2 unrelated patients, thus confirming its natural immunogenicity. We further investigated the neoantigen-specific clones and their T cell receptors (TCRs) via a combination of TCR sequencing, TCR overexpression, functional assays, and single-cell transcriptomics. Our analysis revealed a diverse repertoire of neoantigen-specific clones with both intra- and interpatient TCR similarities. Moreover, 1 dominant clone proved to cross-react with the highly prevalent RAS.Q61R variant. Transcriptome analysis revealed a high association of TCR clones with specific T cell phenotypes in response to cognate melanoma, with neoantigen-specific cells showing an activated and dysfunctional phenotype. Identification of recurrent neoantigens and their reactive TCRs can promote "off-the-shelf" precision immunotherapies, alleviating limitations of personalized treatments.
Keywords: Antigen; Genetics; MHC class 1; Melanoma; Oncology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
