

New genes involved in Angelman syndrome-like: Expanding the genetic spectrum
- PMID: 34653234
- PMCID: PMC8519432
- DOI: 10.1371/journal.pone.0258766
New genes involved in Angelman syndrome-like: Expanding the genetic spectrum
Abstract
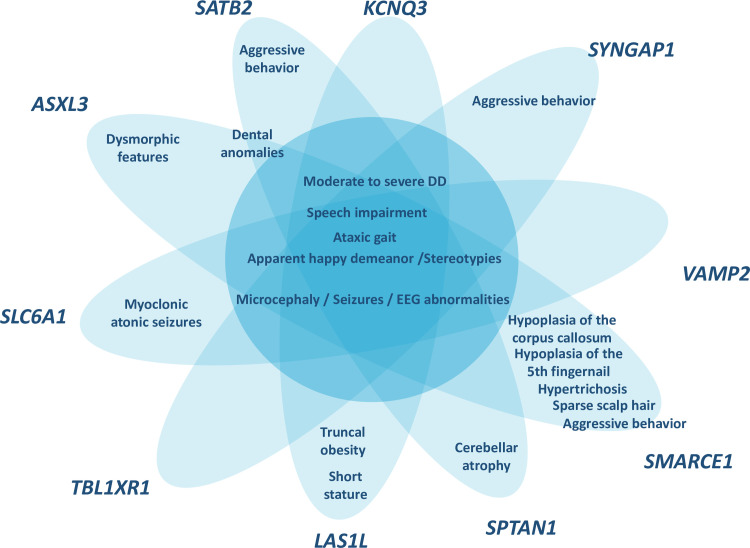
Angelman syndrome (AS) is a neurogenetic disorder characterized by severe developmental delay with absence of speech, happy disposition, frequent laughter, hyperactivity, stereotypies, ataxia and seizures with specific EEG abnormalities. There is a 10-15% of patients with an AS phenotype whose genetic cause remains unknown (Angelman-like syndrome, AS-like). Whole-exome sequencing (WES) was performed on a cohort of 14 patients with clinical features of AS and no molecular diagnosis. As a result, we identified 10 de novo and 1 X-linked pathogenic/likely pathogenic variants in 10 neurodevelopmental genes (SYNGAP1, VAMP2, TBL1XR1, ASXL3, SATB2, SMARCE1, SPTAN1, KCNQ3, SLC6A1 and LAS1L) and one deleterious de novo variant in a candidate gene (HSF2). Our results highlight the wide genetic heterogeneity in AS-like patients and expands the differential diagnosis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Comment in
-
Novel causal genes in Angelman syndrome.Nat Rev Neurol. 2021 Dec;17(12):726. doi: 10.1038/s41582-021-00587-5. Nat Rev Neurol. 2021. PMID: 34732830 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
