

Enhanced prime editing systems by manipulating cellular determinants of editing outcomes
- PMID: 34653350
- PMCID: PMC8584034
- DOI: 10.1016/j.cell.2021.09.018
Enhanced prime editing systems by manipulating cellular determinants of editing outcomes
Abstract
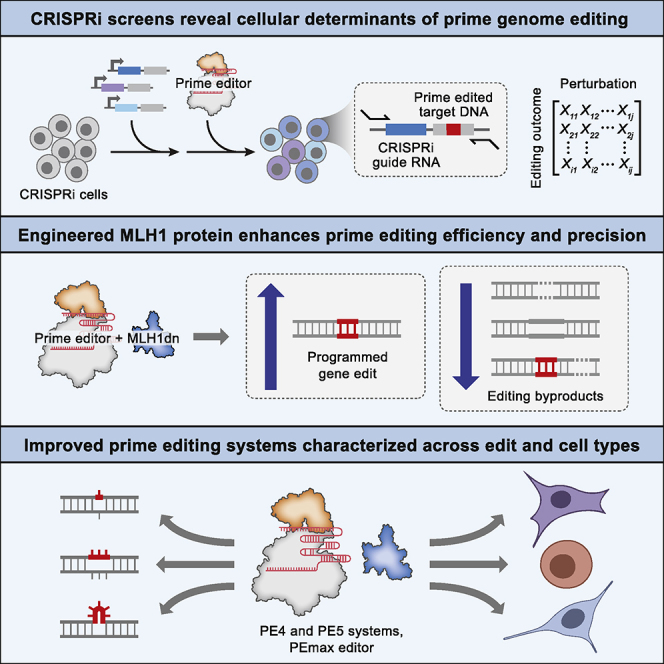
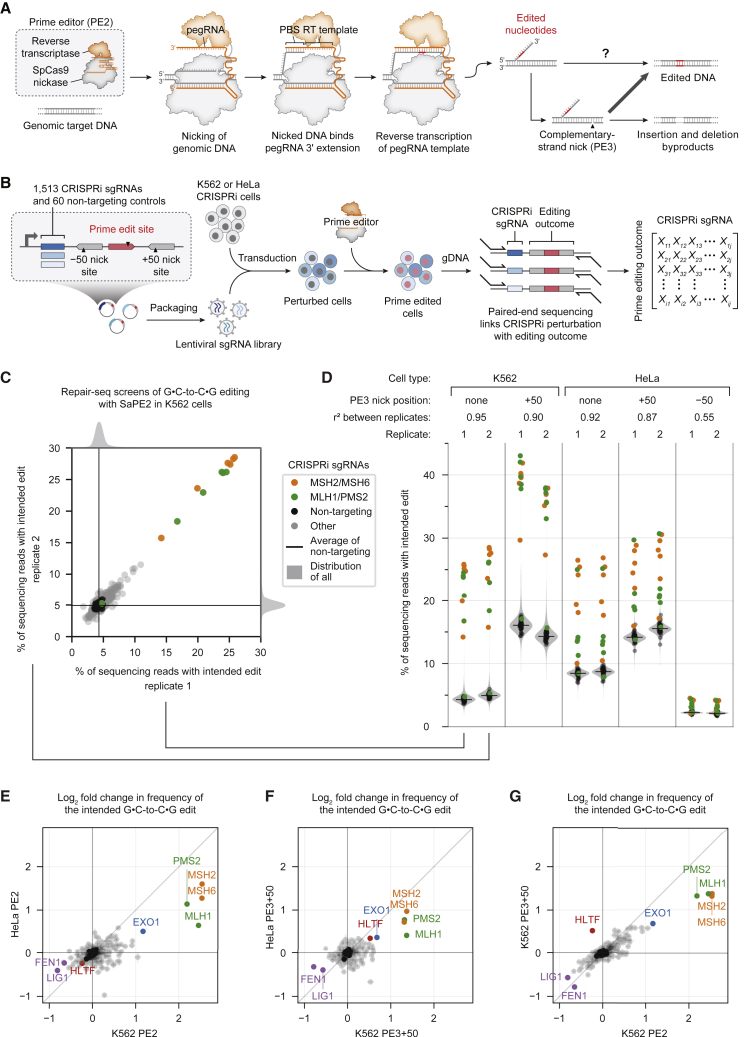
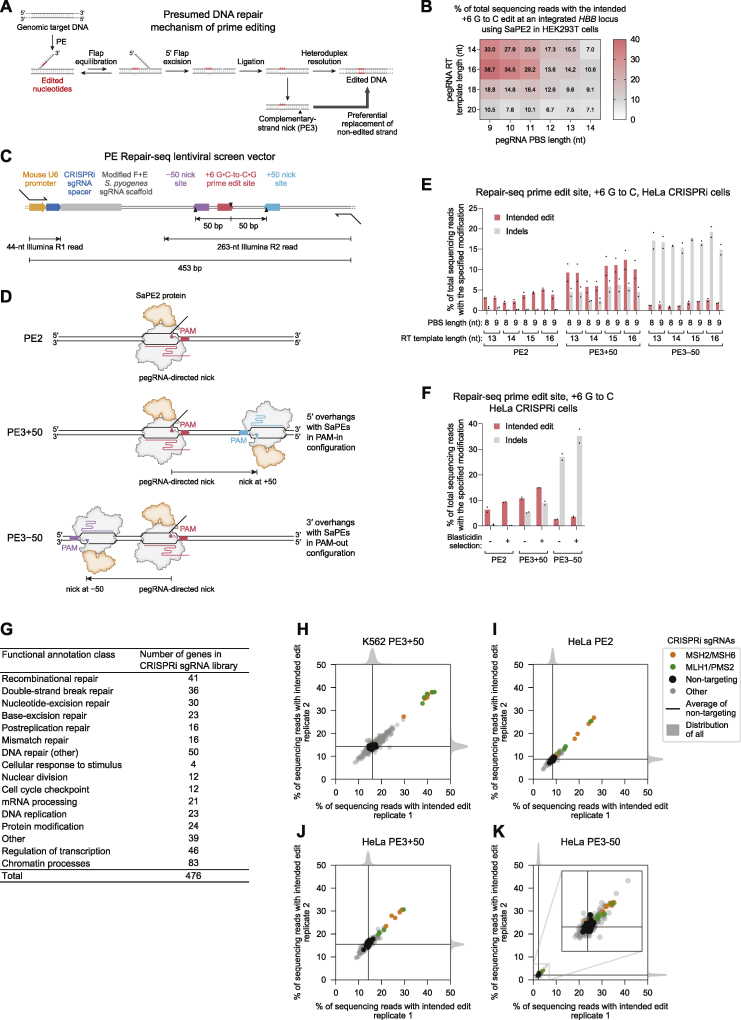
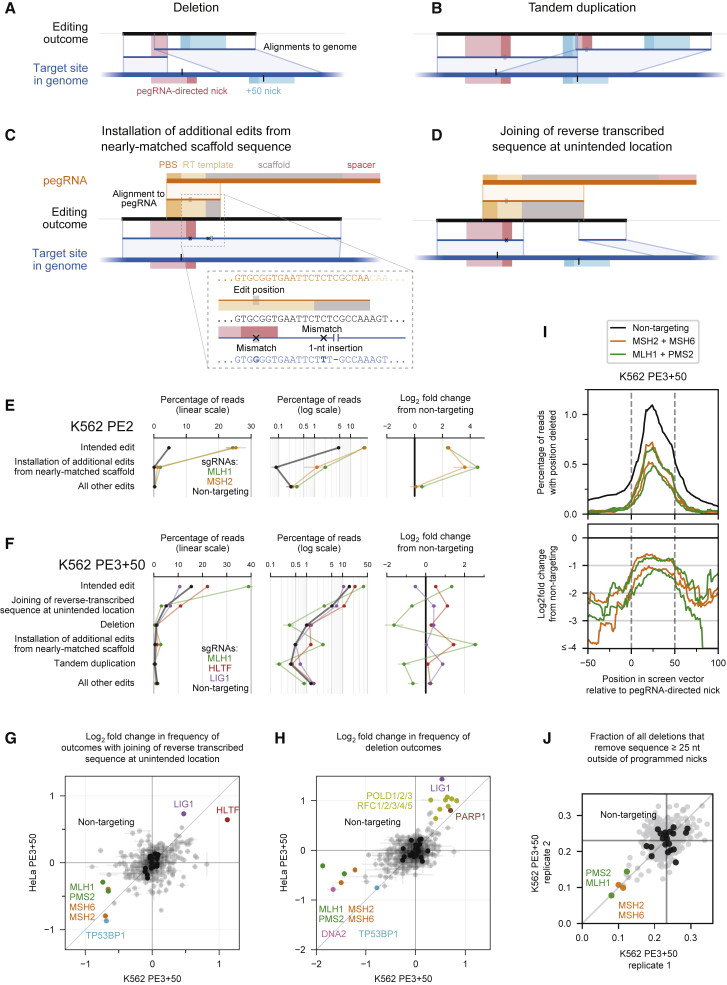
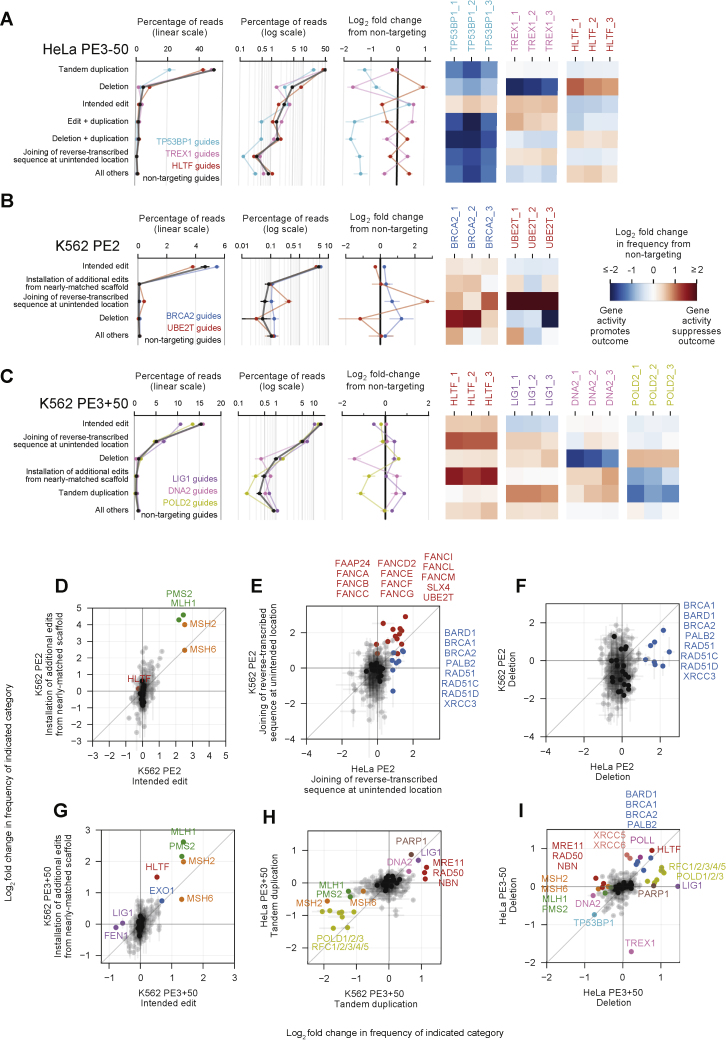
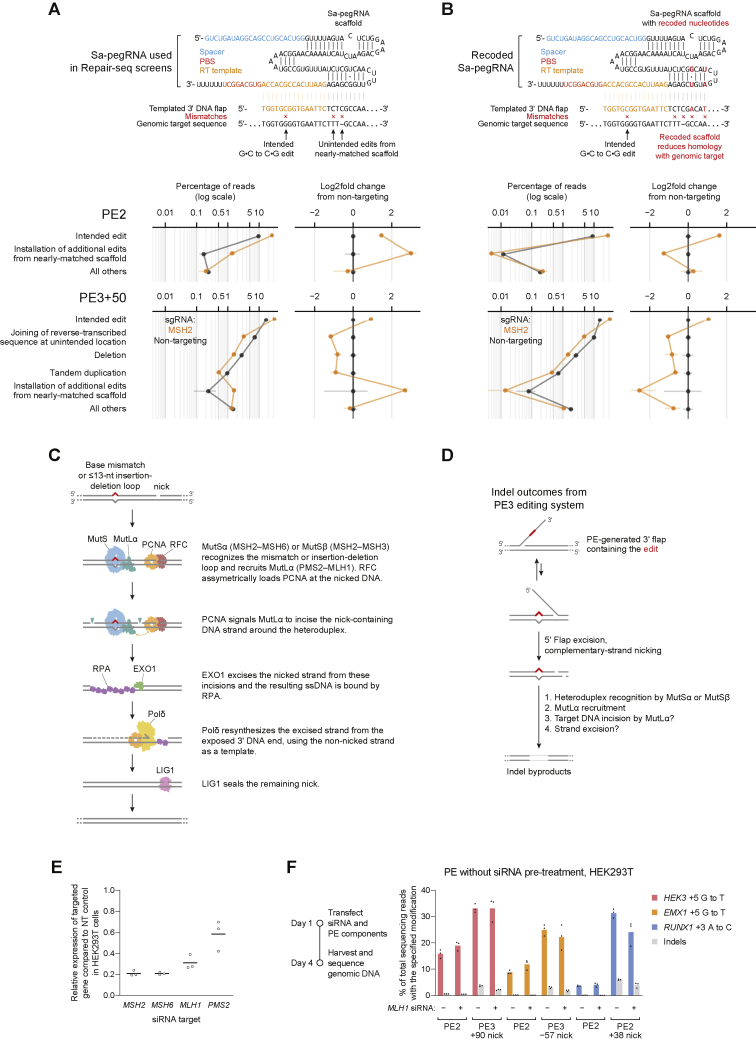
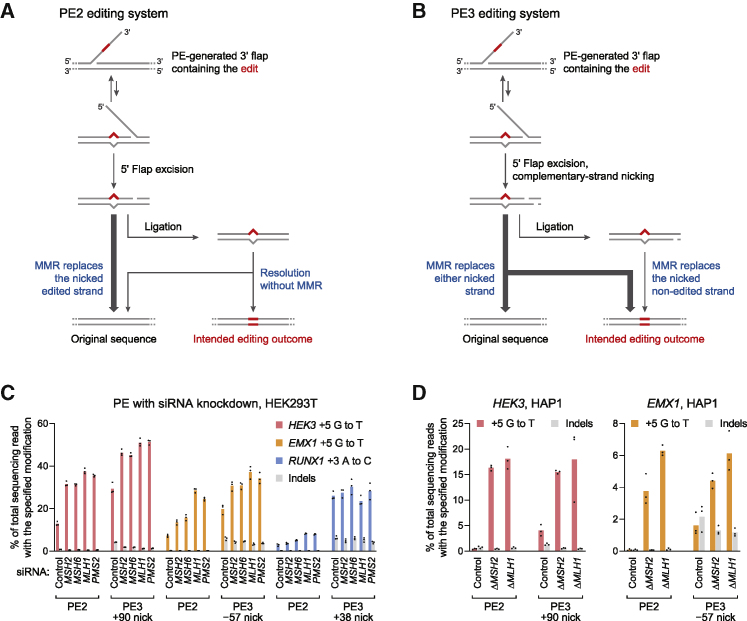
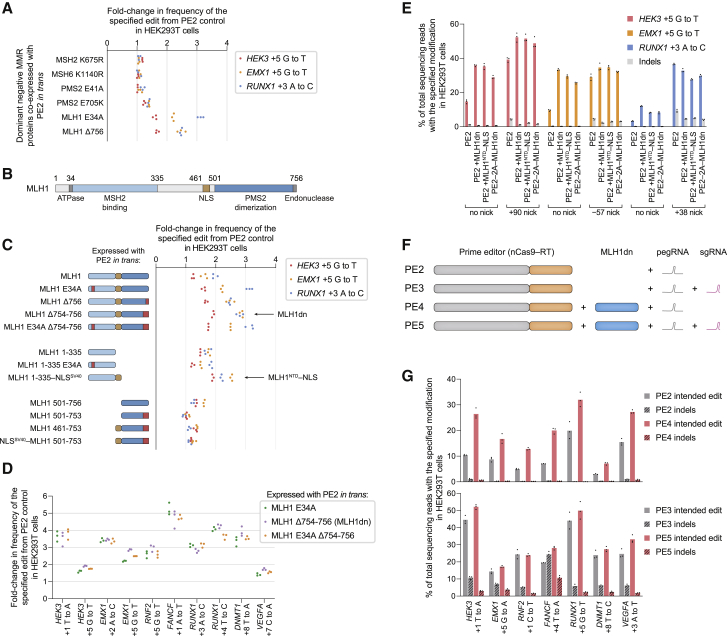
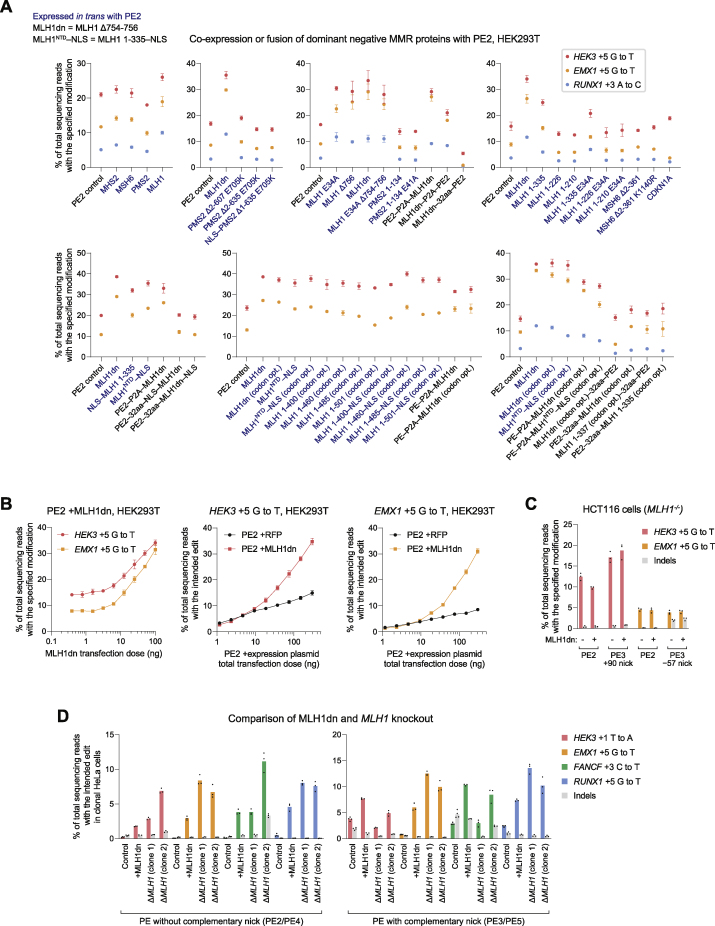
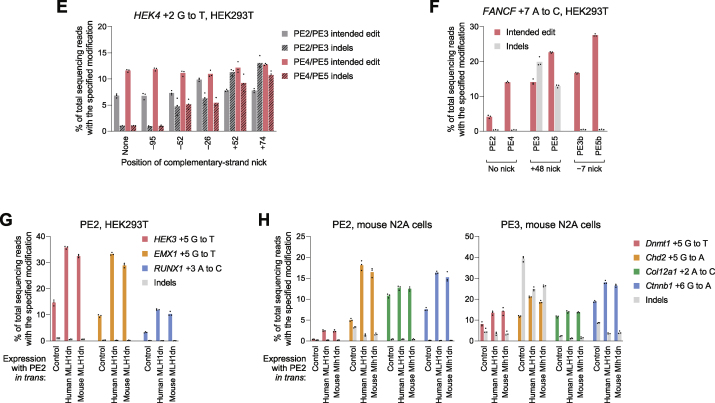
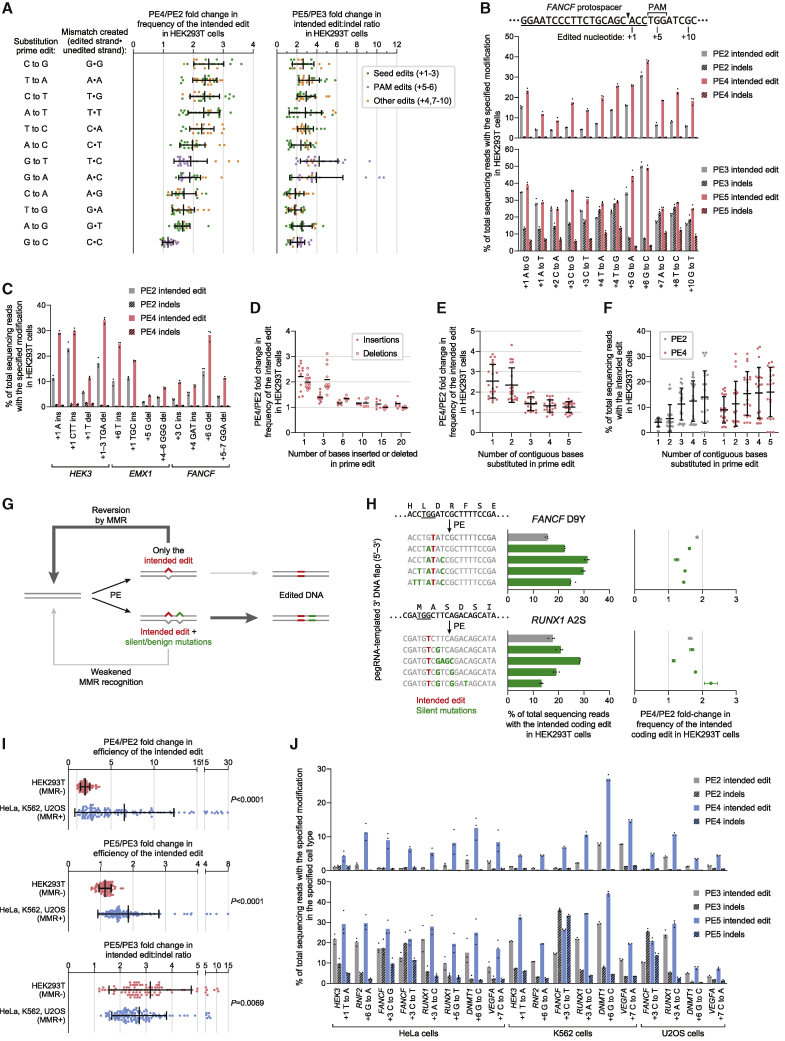
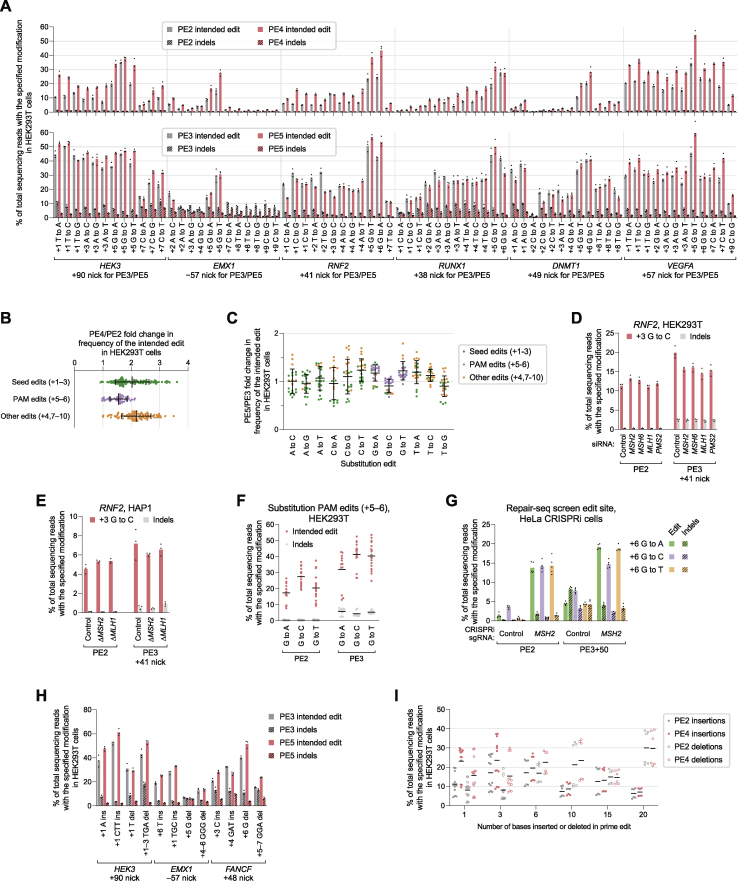
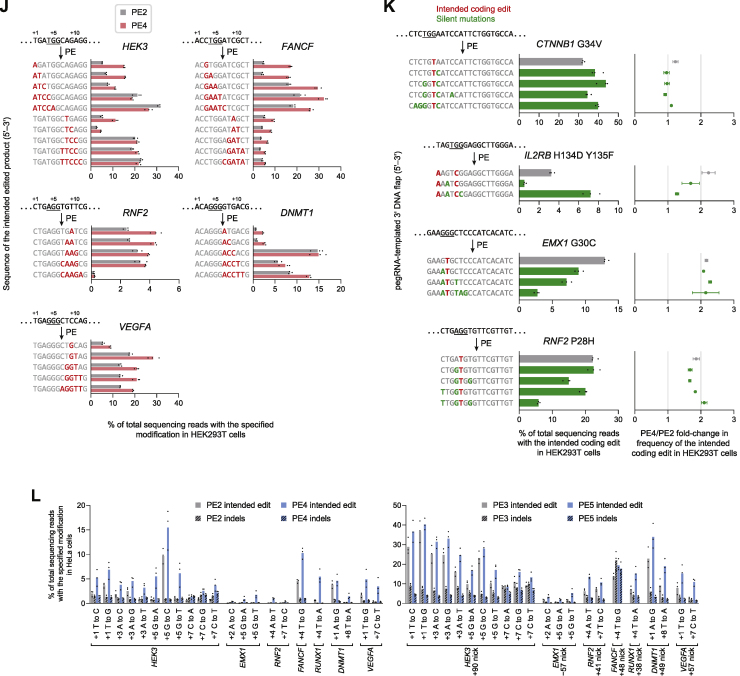
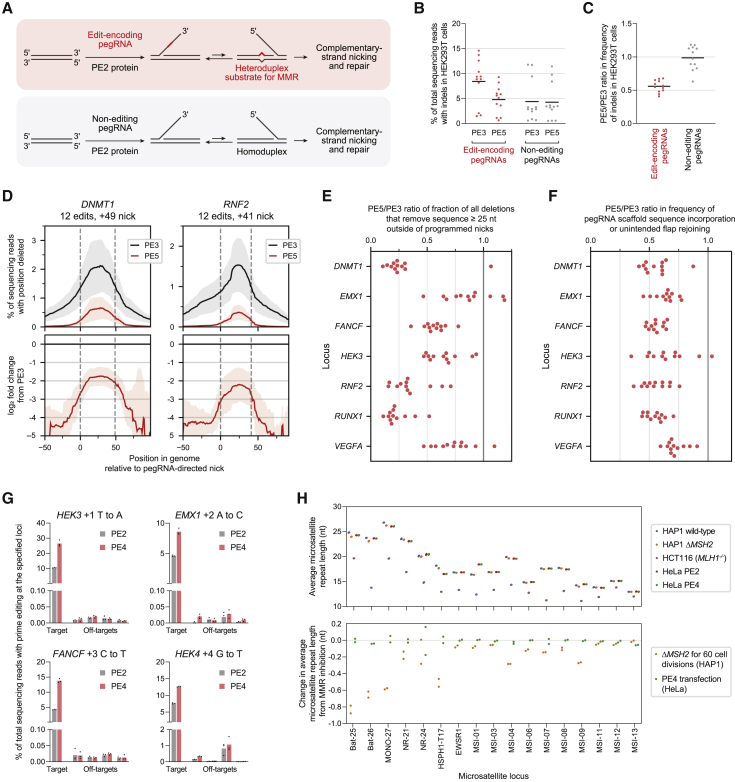
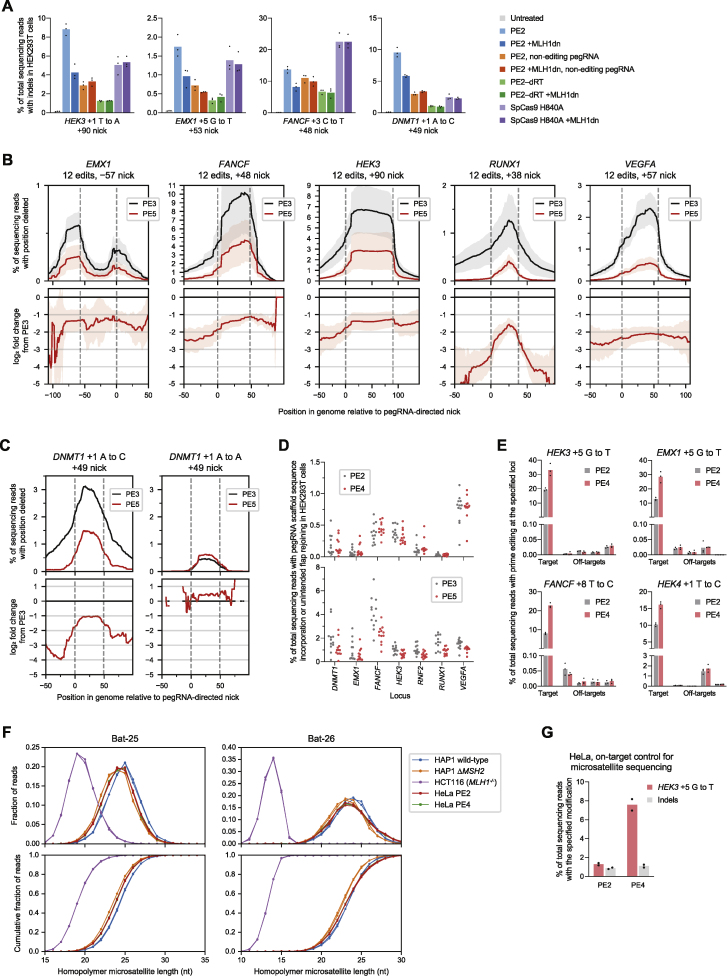
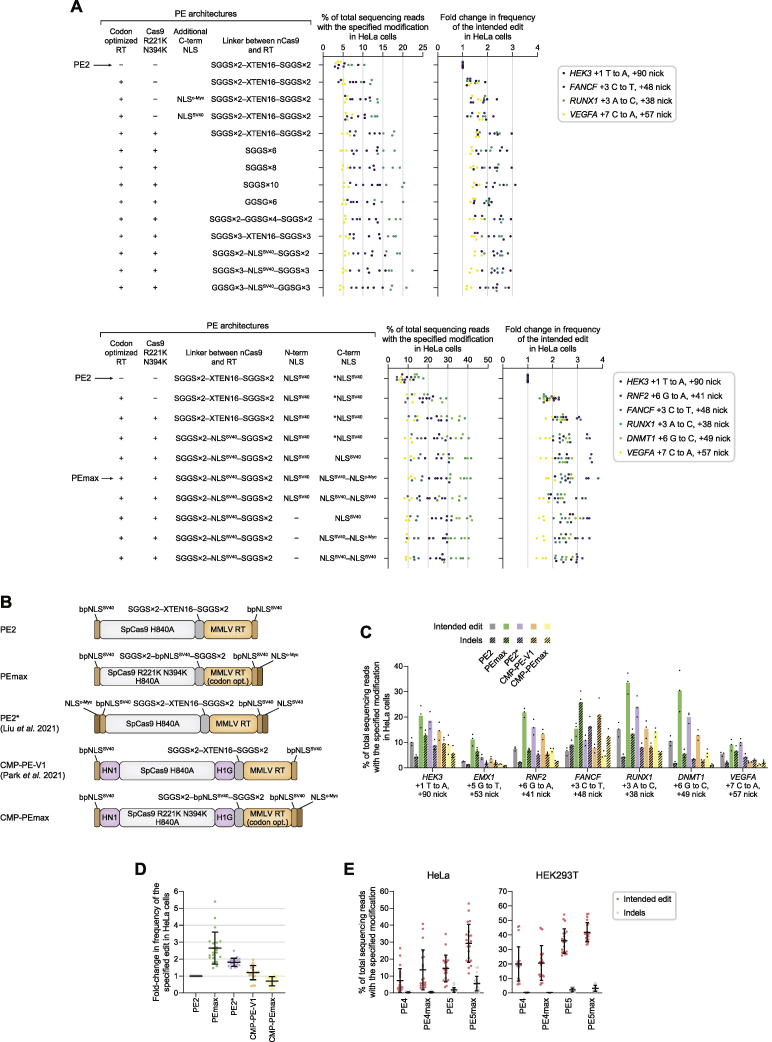
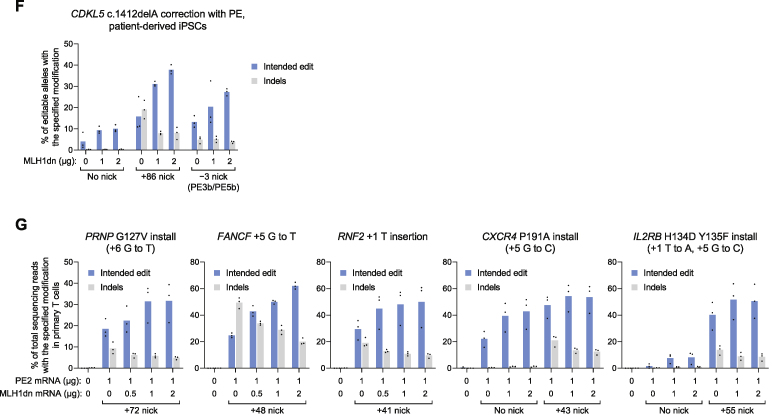
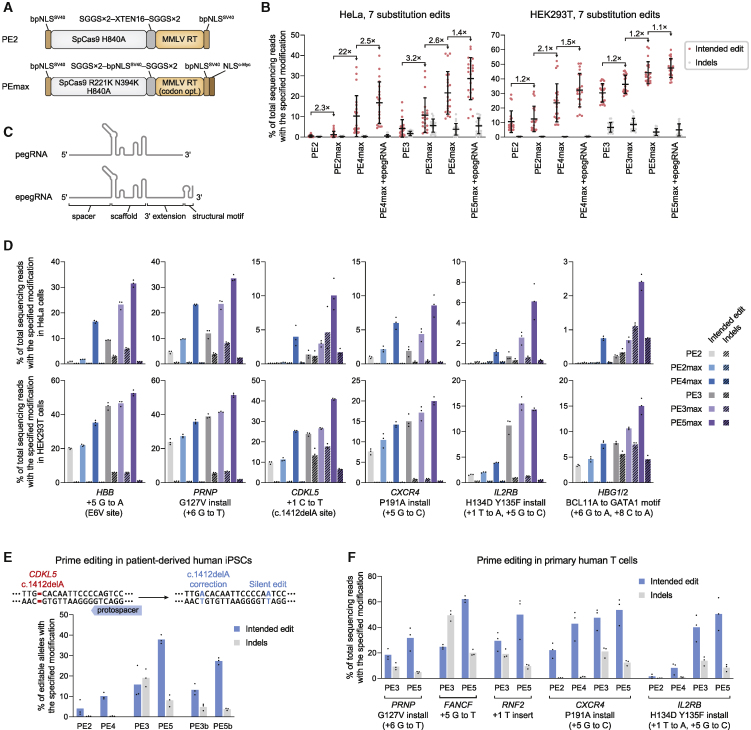
While prime editing enables precise sequence changes in DNA, cellular determinants of prime editing remain poorly understood. Using pooled CRISPRi screens, we discovered that DNA mismatch repair (MMR) impedes prime editing and promotes undesired indel byproducts. We developed PE4 and PE5 prime editing systems in which transient expression of an engineered MMR-inhibiting protein enhances the efficiency of substitution, small insertion, and small deletion prime edits by an average 7.7-fold and 2.0-fold compared to PE2 and PE3 systems, respectively, while improving edit/indel ratios by 3.4-fold in MMR-proficient cell types. Strategic installation of silent mutations near the intended edit can enhance prime editing outcomes by evading MMR. Prime editor protein optimization resulted in a PEmax architecture that enhances editing efficacy by 2.8-fold on average in HeLa cells. These findings enrich our understanding of prime editing and establish prime editing systems that show substantial improvement across 191 edits in seven mammalian cell types.
Keywords: CRISPR-Cas9; Repair-seq; genome editing; mismatch repair; prime editing.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests P.J.C., J.A.H., B.A., and D.R.L. have filed patent applications on aspects of this work through their respective institutions. J.A.H. is a consultant for Tessera Therapeutics. P.-F.C. is currently an employee of Tessera Therapeutics. B.A. was a member of a ThinkLab Advisory Board for, and holds equity in, Celsius Therapeutics. D.R.L. is a consultant and equity holder of Beam Therapeutics, Prime Medicine, Pairwise Plants, and Chroma Medicine.
Figures
Comment in
-
Illuminating the path to DNA repair.Cell. 2021 Oct 28;184(22):5503-5505. doi: 10.1016/j.cell.2021.10.005. Cell. 2021. PMID: 34715020
-
Improving CRISPR tools by elucidating DNA repair.Nat Biotechnol. 2021 Dec;39(12):1512-1514. doi: 10.1038/s41587-021-01149-2. Nat Biotechnol. 2021. PMID: 34873327 No abstract available.
References
-
- Amato A., Cappabianca M.P., Perri M., Zaghis I., Grisanti P., Ponzini D., Di Biagio P. Interpreting elevated fetal hemoglobin in pathology and health at the basic laboratory level: new and known γ- gene mutations associated with hereditary persistence of fetal hemoglobin. Int. J. Lab. Hematol. 2014;36:13–19. - PubMed
-
- Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime. Nat. Biotechnol. 2020;38:824–844. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL156647/HL/NHLBI NIH HHS/United States
- R35 GM138167/GM/NIGMS NIH HHS/United States
- P30 CA072720/CA/NCI NIH HHS/United States
- RM1 HG009490/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R35 GM118062/GM/NIGMS NIH HHS/United States
- U01 AI142756/AI/NIAID NIH HHS/United States
- U54 HD090255/HD/NICHD NIH HHS/United States
- P50 HD105351/HD/NICHD NIH HHS/United States
- P01 CA065493/CA/NCI NIH HHS/United States
- R01 EB031172/EB/NIBIB NIH HHS/United States
- R01 AR063070/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
