

CHD1L augments autophagy-mediated migration of hepatocellular carcinoma through targeting ZKSCAN3
- PMID: 34654797
- PMCID: PMC8520006
- DOI: 10.1038/s41419-021-04254-x
CHD1L augments autophagy-mediated migration of hepatocellular carcinoma through targeting ZKSCAN3
Abstract
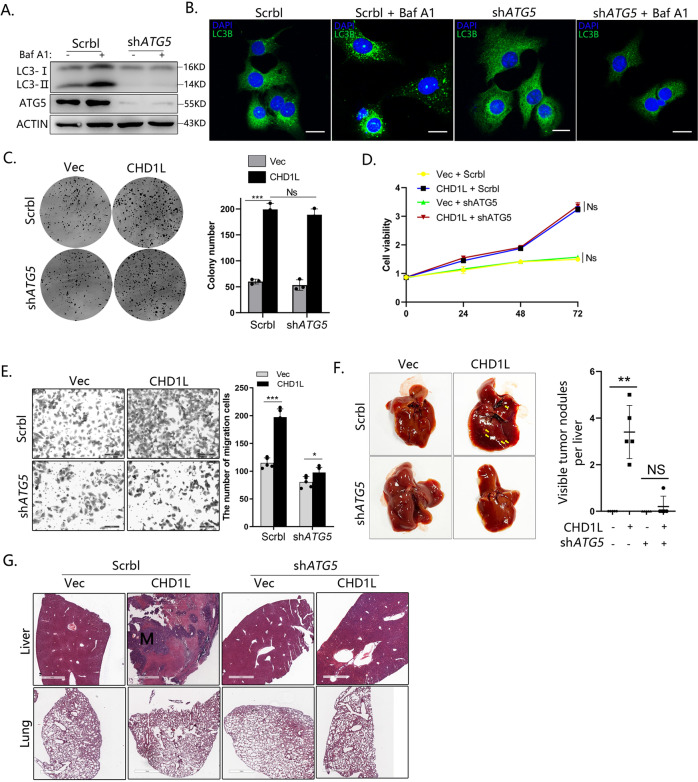
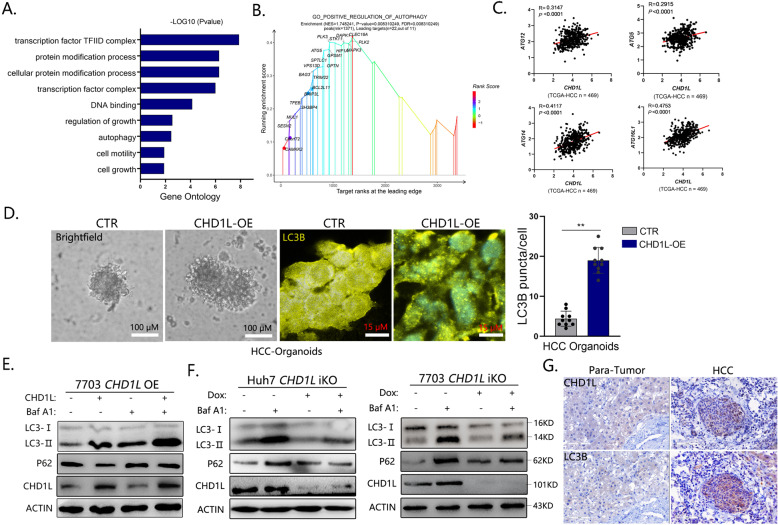
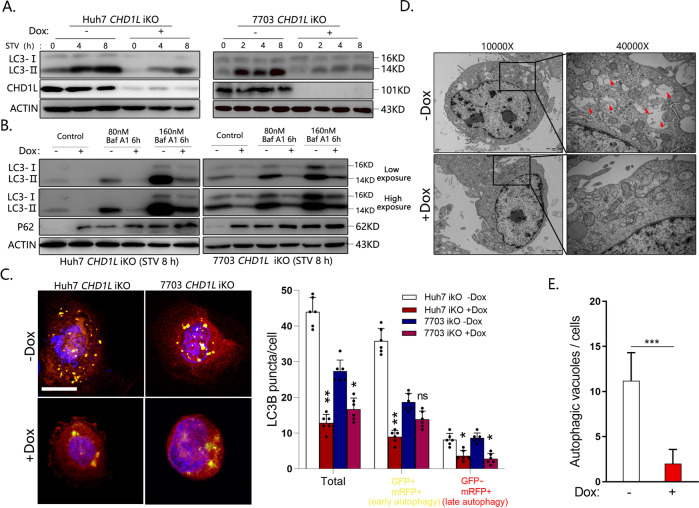
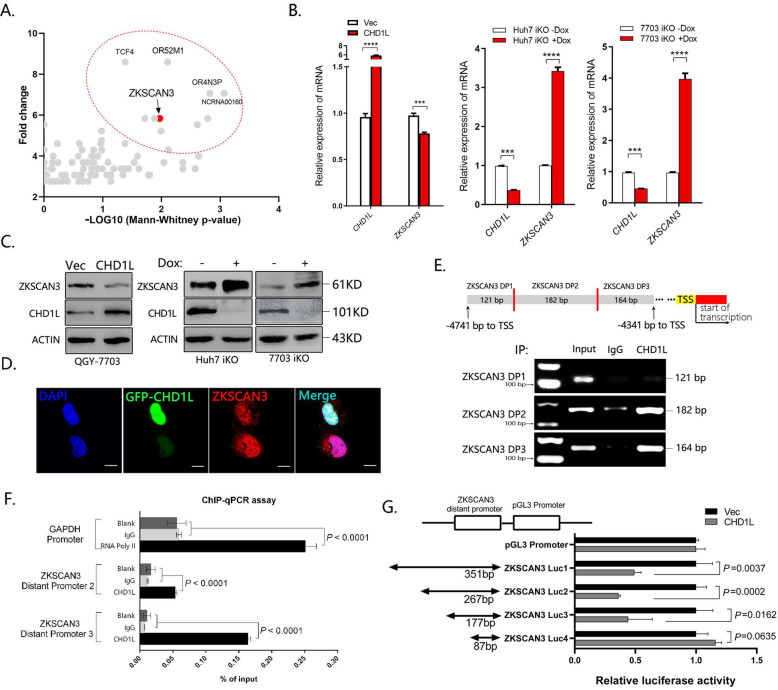
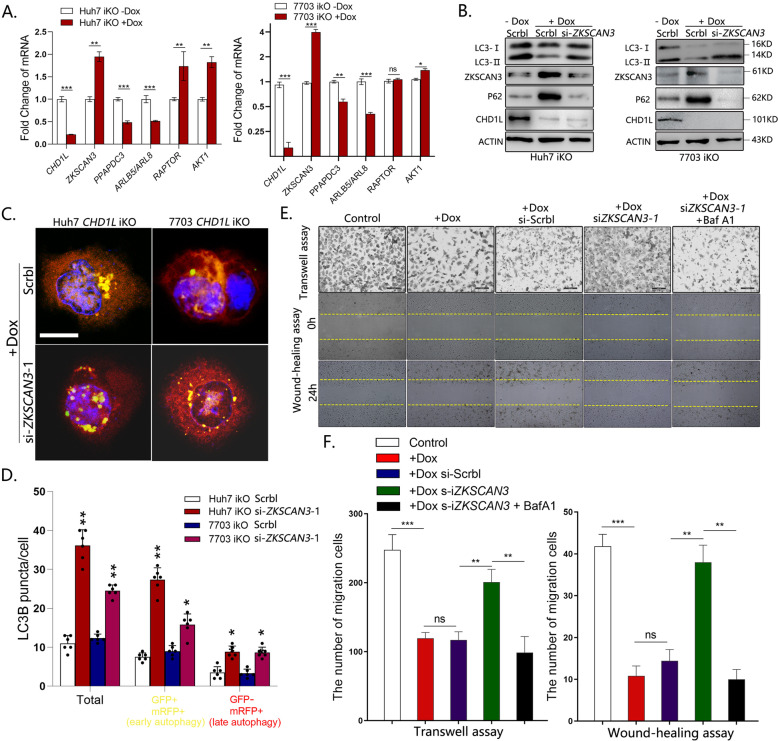
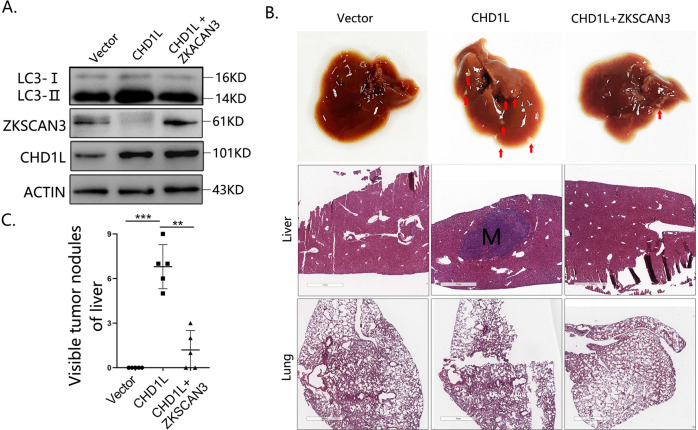
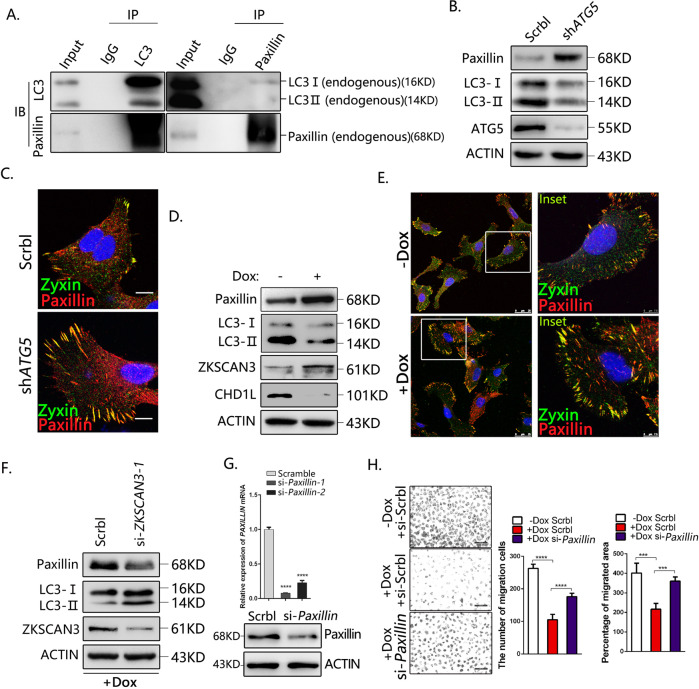
Autophagy is an important biological process in normal cells. However, how it affects tumor progression still remains poorly understood. Herein, we demonstrated that the oncogenic protein Chromodomain-helicase-DNA-binding-protein 1-like gene (CHD1L) might promote HCC cells migration and metastasis through autophagy. CHD1L could bind to the promotor region of Zinc finger with KRAB and SCAN domain 3 (ZKSCAN3), a pivotal autophagy suppressor, and inhibit its transcription. We established inducible CHD1L conditional knockout cell line (CHD1L-iKO cell) and found that the deletion of CHD1L significantly increased ZKSCAN3 expression both at mRNA and protein level. Deletion of CHD1L impaired the autophagic flux and migration of HCC cells, while specifically inhibiting ZKSCAN3 blocked these effects. Further exploration demonstrated that the enhanced tumor cell migration and metastasis induced by CHD1L was mediated through ZKSCAN3-induced autophagic degradation of Paxillin. In summary, we have characterized a previously unknown function of CHD1L in regulating tumor migration via ZKSCAN3-mediated autophagy in HCC. Further inhibition of CHD1L and its downstream autophagy signaling might shed new light on cancer therapeutics.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9:1167–81. doi: 10.1158/2159-8290.CD-19-0292. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
