

Extracellular Histones Bind Vascular Glycosaminoglycans and Inhibit the Anti-Inflammatory Function of Antithrombin
- PMID: 34655467
- PMCID: PMC8552502
- DOI: 10.33594/000000438
Extracellular Histones Bind Vascular Glycosaminoglycans and Inhibit the Anti-Inflammatory Function of Antithrombin
Abstract
Background/aims: Binding of histones to molecular pattern recognition receptors on endothelial cells and leukocytes provokes proinflammatory responses and promotes activation of coagulation. Histones also bind therapeutic heparins, thereby neutralizing their anticoagulant functions. The aim of this study was to test the hypothesis that histones can interact with the antithrombin (AT)-binding vascular glycosaminoglycans (GAGs) to induce inflammation and inhibit the anti-inflammatory function of AT.
Methods: We evaluated the heparin-binding function of histones by an AT-dependent protease-inhibition assay. Furthermore, we treated endothelial cells with histones in the absence and presence of AT and monitored cellular phenotypes employing established signaling assays.
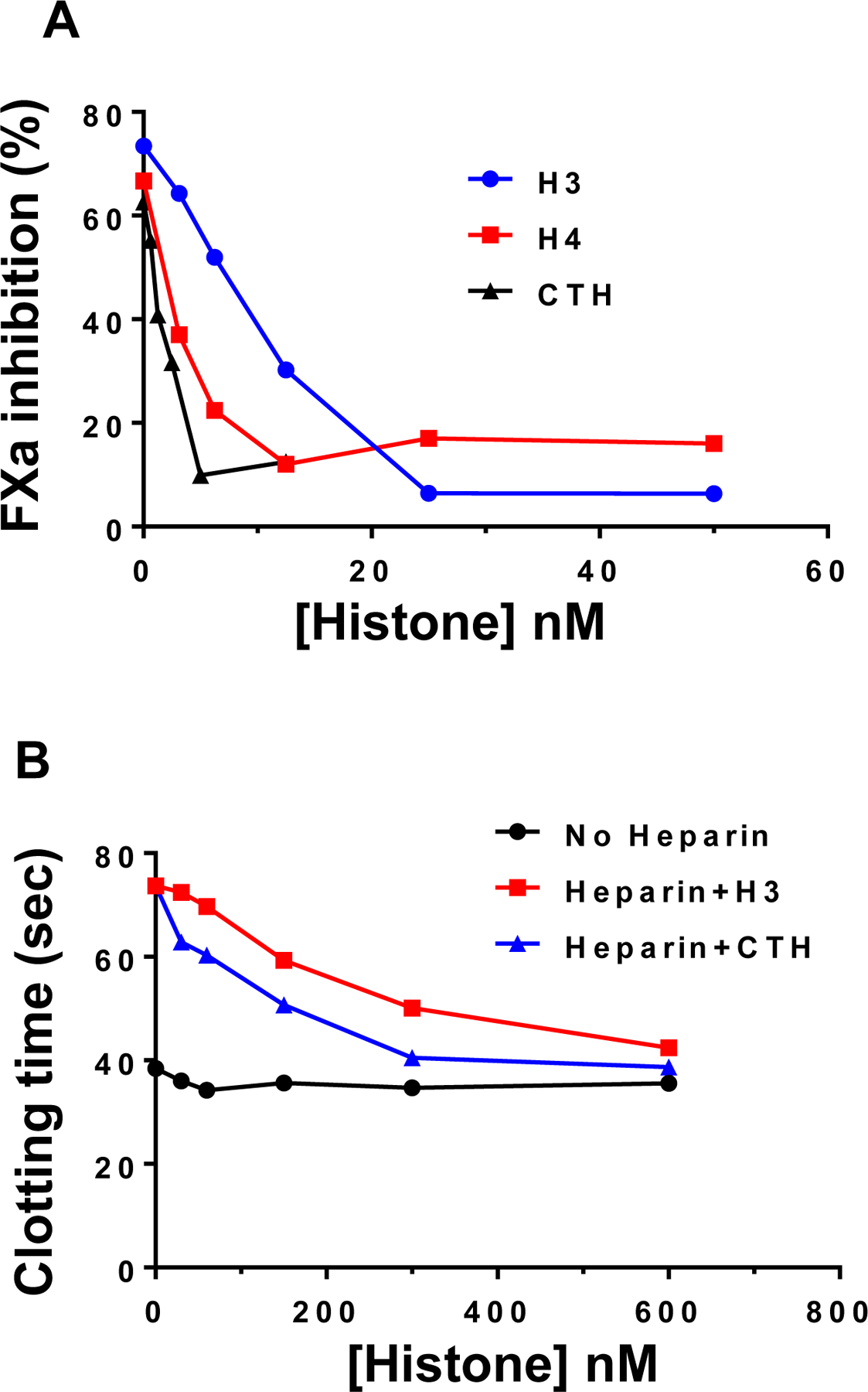
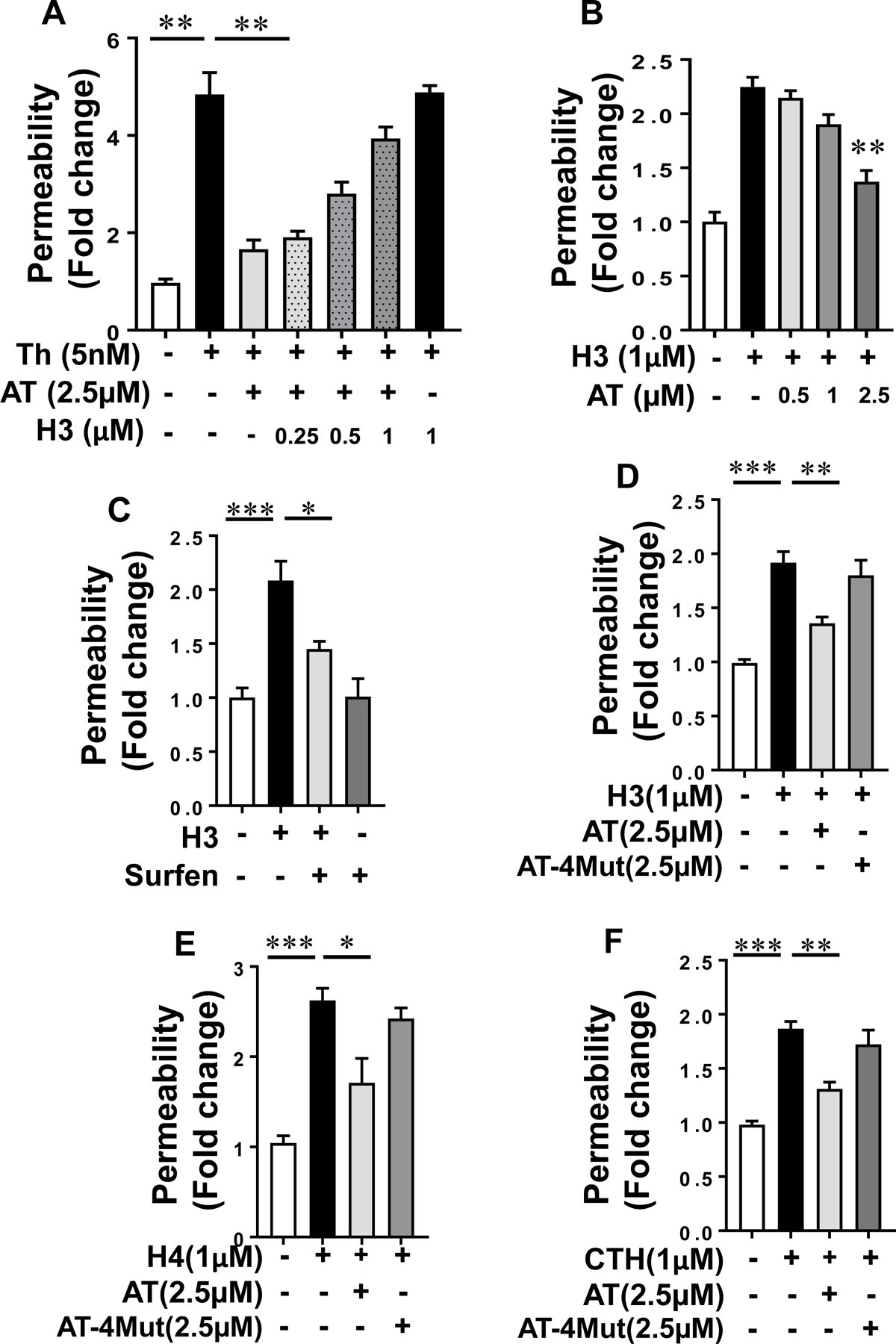
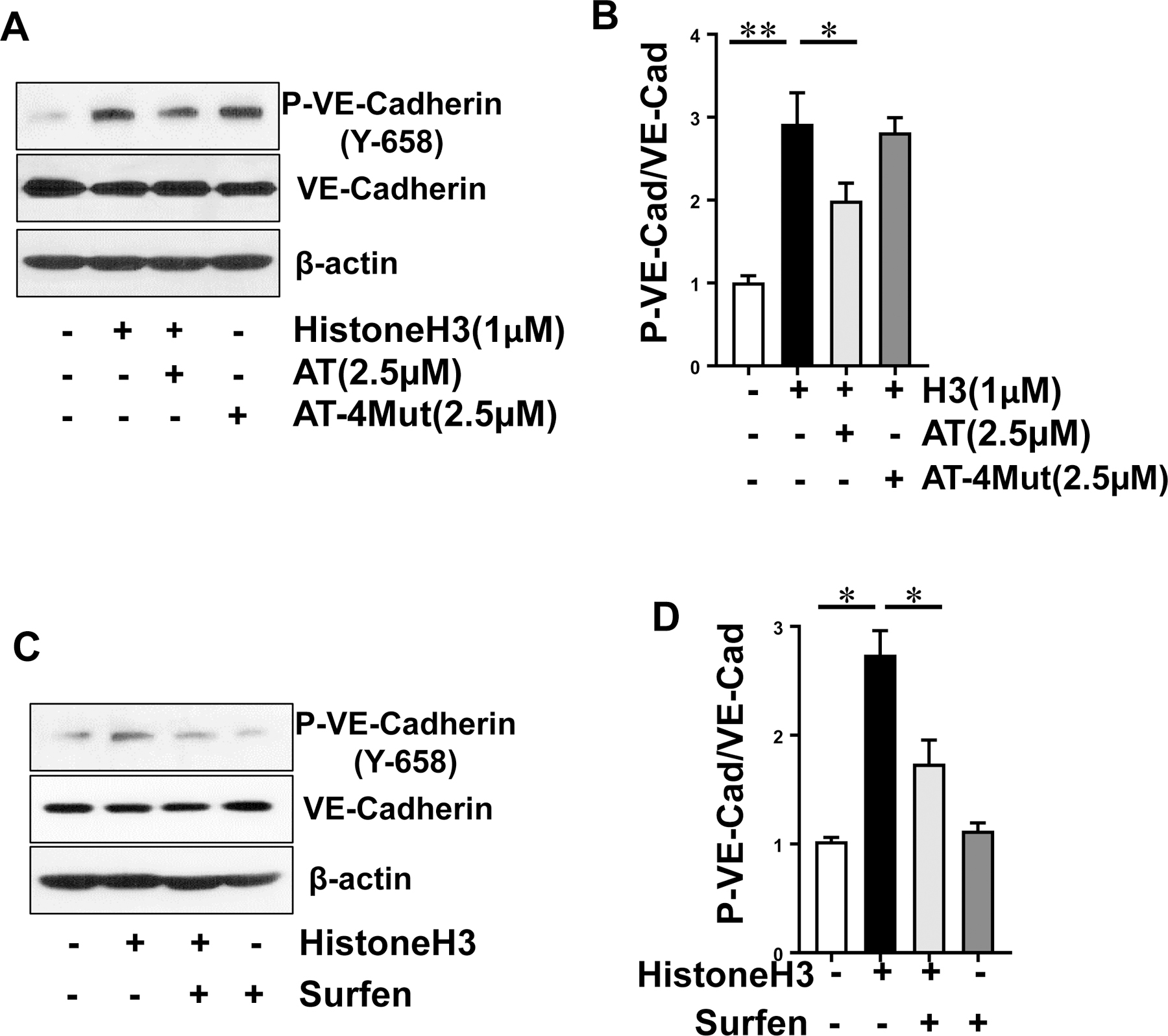
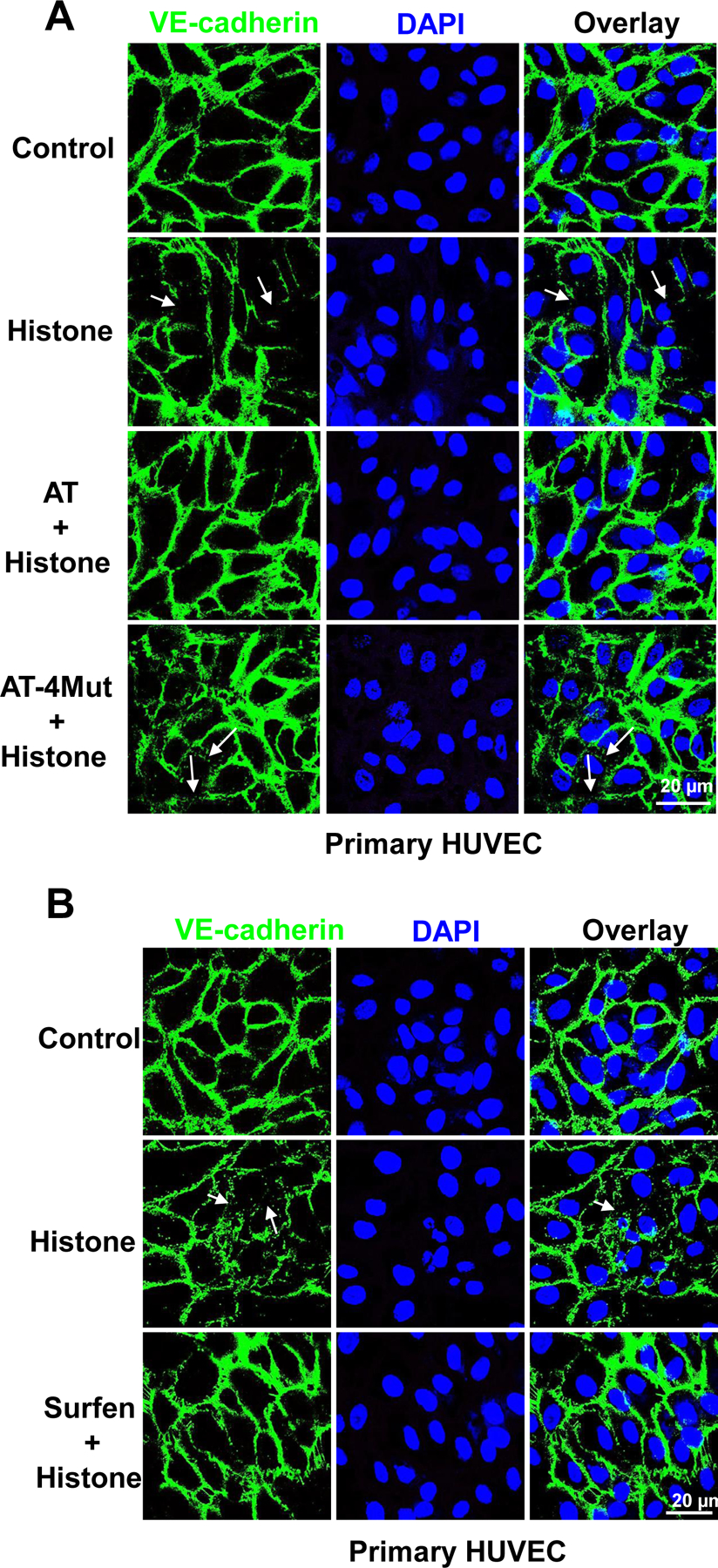
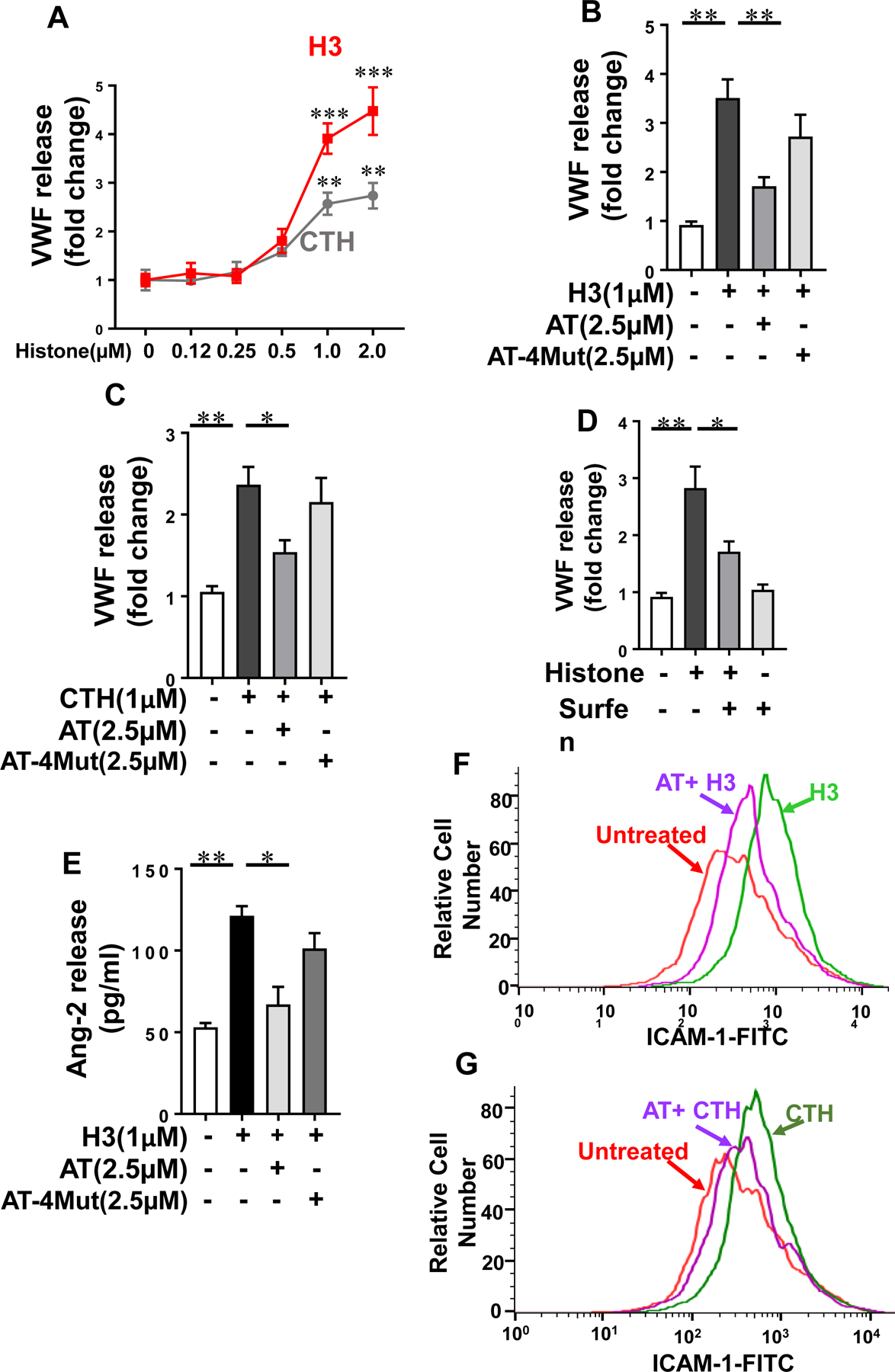
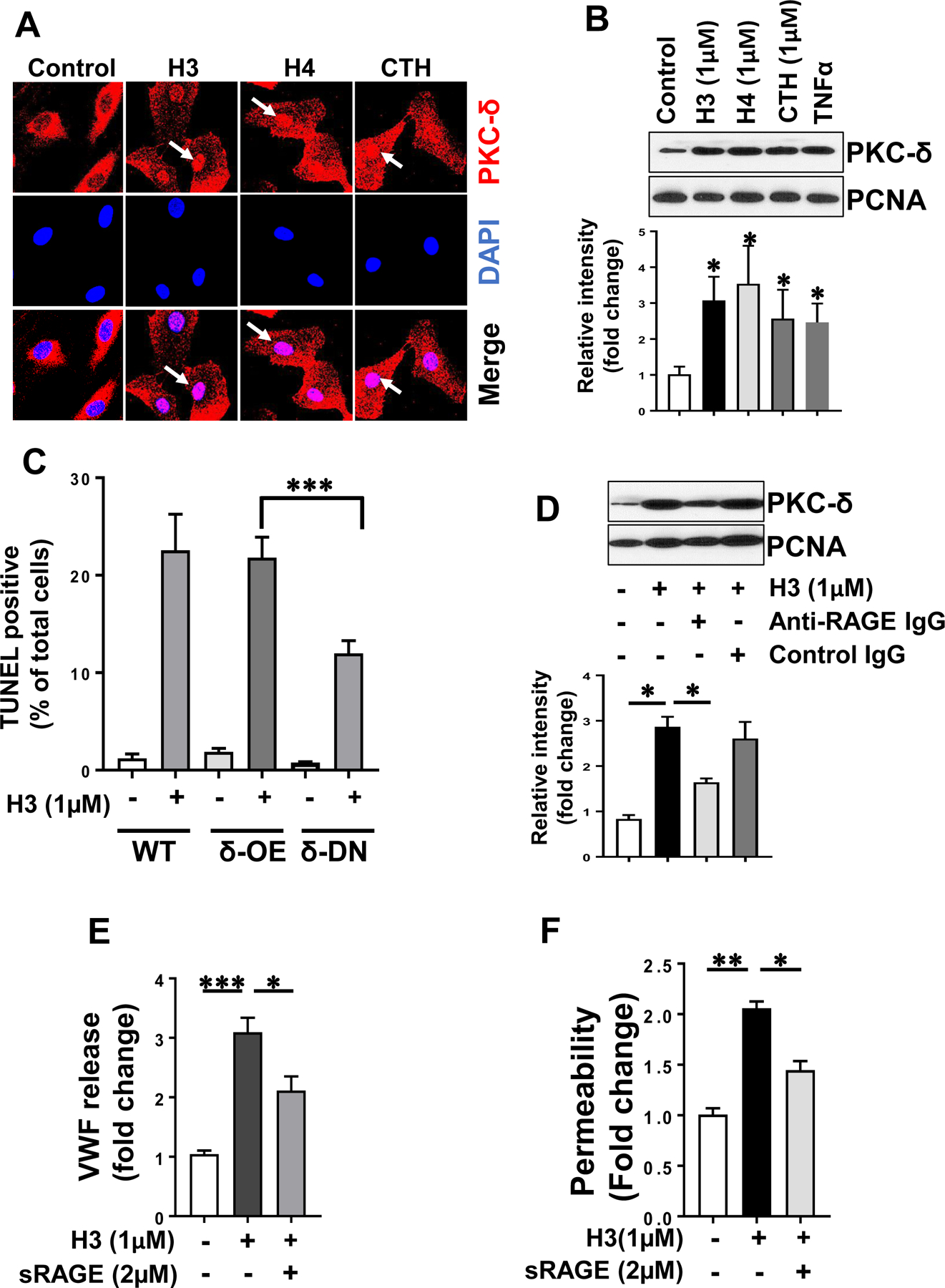
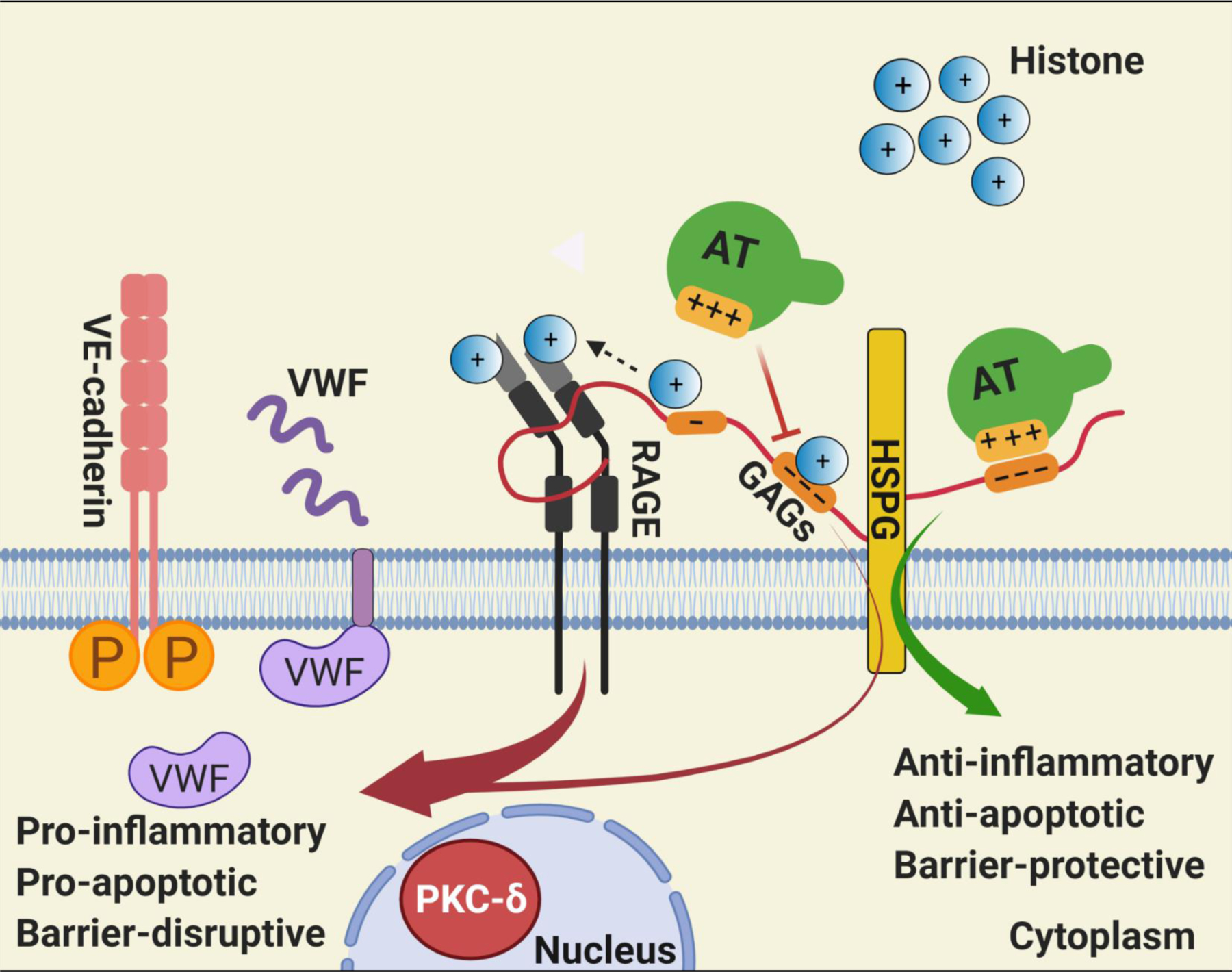
Results: Histones neutralized AT-dependent anticoagulant function of heparin in both purified protease-inhibition and plasma-based assays. Histones also disrupted endothelial cell barrier-permeability function by a GAG-dependent mechanism as evidenced by the GAG-antagonist, surfen, abrogating their disruptive effects. Further studies revealed histones and AT compete for overlapping binding-sites on GAGs, thus increasing concentrations of one protein abrogated effects of the other. Histones elicited proapoptotic effects by inducing nuclear localization of PKC-δ in endothelial cells and barrier-disruptive effects by destabilizing VE-cadherin, which were inhibited by AT, but not by a D-helix mutant of AT incapable of interacting with GAGs. Finally, histones induced release of Weibel-Palade body contents, VWF and angiopoietin-2, and promoted expression of cell adhesion molecules on endothelial cells, which were all downregulated by AT but not by D-helix mutant of AT.
Conclusion: We conclude that histones and AT compete for overlapping binding sites on vascular GAGs to modulate coagulation and inflammation.
Keywords: Histones; Antithrombin; Glycosaminoglycans; Signaling.
© Copyright by the Author(s). Published by Cell Physiol Biochem Press.
Conflict of interest statement
The authors declare no conflict of interests exists.
Figures
References
-
- Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet 2009;43:559–599. - PubMed
-
- Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double-edged swords in immunothrombosis. J Thromb Haemost 2015;Suppl 1:S82–91. - PubMed
-
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 2004;303:1532–5153. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
