

Cofilin1 oxidation links oxidative distress to mitochondrial demise and neuronal cell death
- PMID: 34657120
- PMCID: PMC8520533
- DOI: 10.1038/s41419-021-04242-1
Cofilin1 oxidation links oxidative distress to mitochondrial demise and neuronal cell death
Abstract
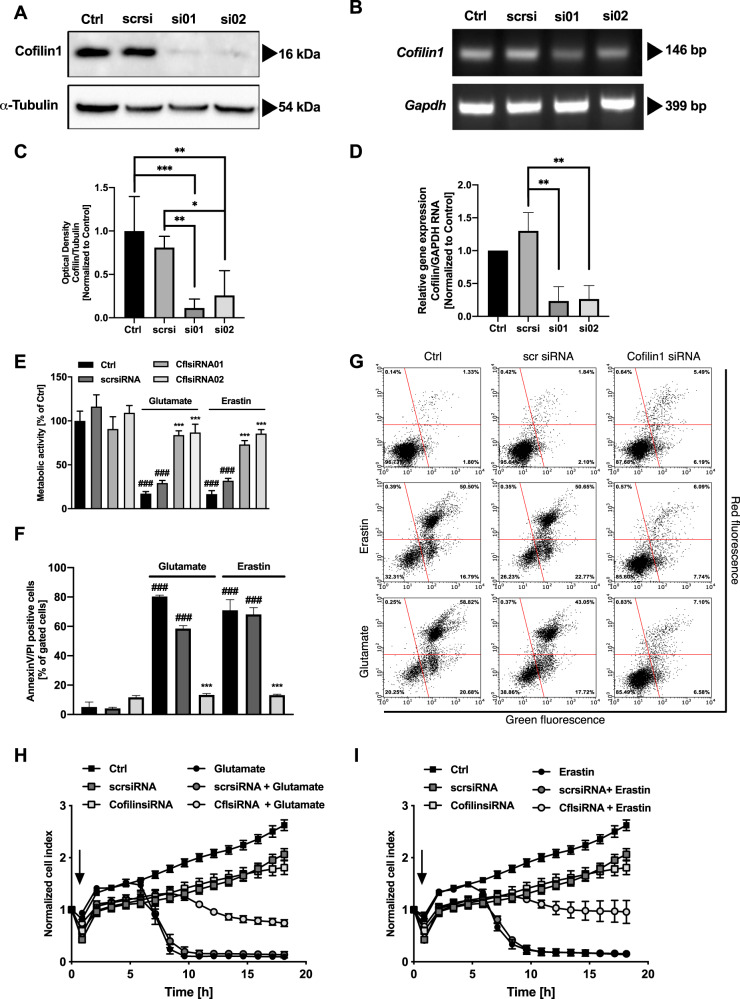
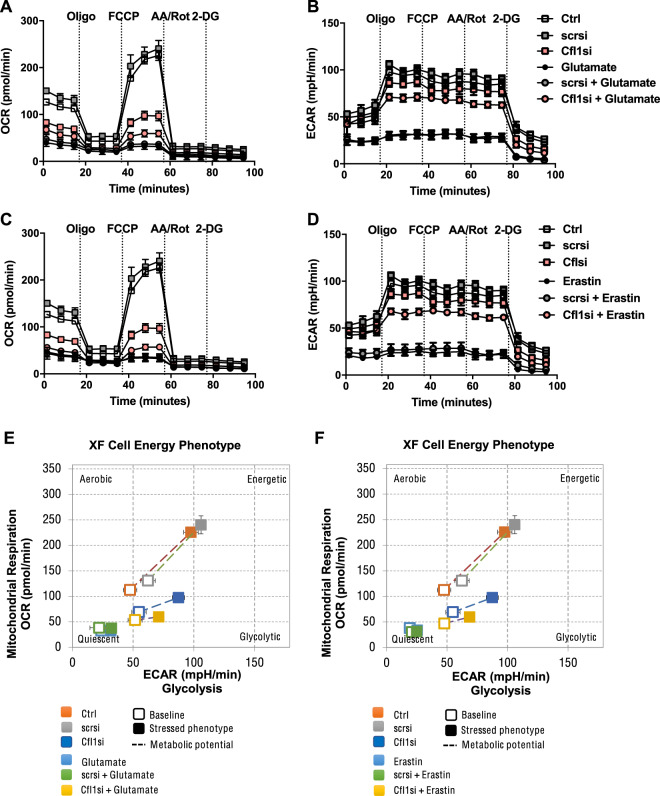
Many cell death pathways, including apoptosis, regulated necrosis, and ferroptosis, are relevant for neuronal cell death and share common mechanisms such as the formation of reactive oxygen species (ROS) and mitochondrial damage. Here, we present the role of the actin-regulating protein cofilin1 in regulating mitochondrial pathways in oxidative neuronal death. Cofilin1 deletion in neuronal HT22 cells exerted increased mitochondrial resilience, assessed by quantification of mitochondrial ROS production, mitochondrial membrane potential, and ATP levels. Further, cofilin1-deficient cells met their energy demand through enhanced glycolysis, whereas control cells were metabolically impaired when challenged by ferroptosis. Further, cofilin1 was confirmed as a key player in glutamate-mediated excitotoxicity and associated mitochondrial damage in primary cortical neurons. Using isolated mitochondria and recombinant cofilin1, we provide a further link to toxicity-related mitochondrial impairment mediated by oxidized cofilin1. Our data revealed that the detrimental impact of cofilin1 on mitochondria depends on the oxidation of cysteine residues at positions 139 and 147. Overall, our findings show that cofilin1 acts as a redox sensor in oxidative cell death pathways of ferroptosis, and also promotes glutamate excitotoxicity. Protective effects by cofilin1 inhibition are particularly attributed to preserved mitochondrial integrity and function. Thus, interfering with the oxidation and pathological activation of cofilin1 may offer an effective therapeutic strategy in neurodegenerative diseases.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
