

Novel Corneal Protein Biomarker Candidates Reveal Iron Metabolic Disturbance in High Myopia Eyes
- PMID: 34660571
- PMCID: PMC8517150
- DOI: 10.3389/fcell.2021.689917
Novel Corneal Protein Biomarker Candidates Reveal Iron Metabolic Disturbance in High Myopia Eyes
Abstract
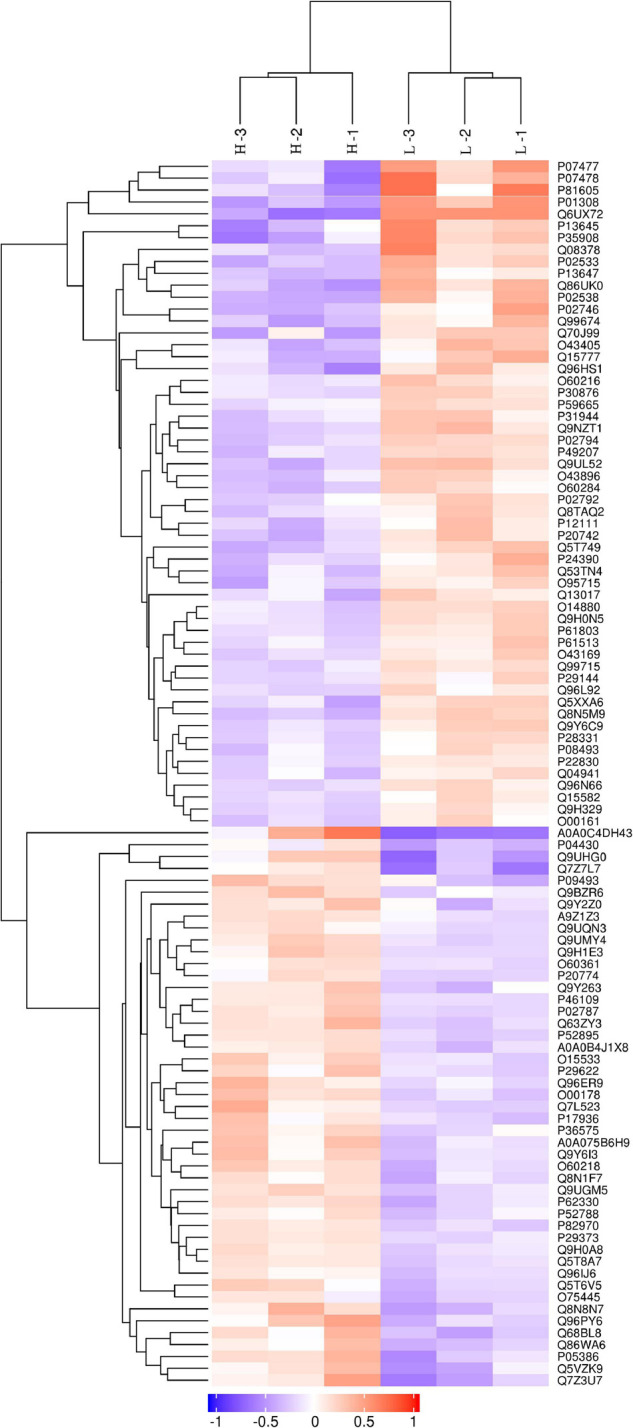
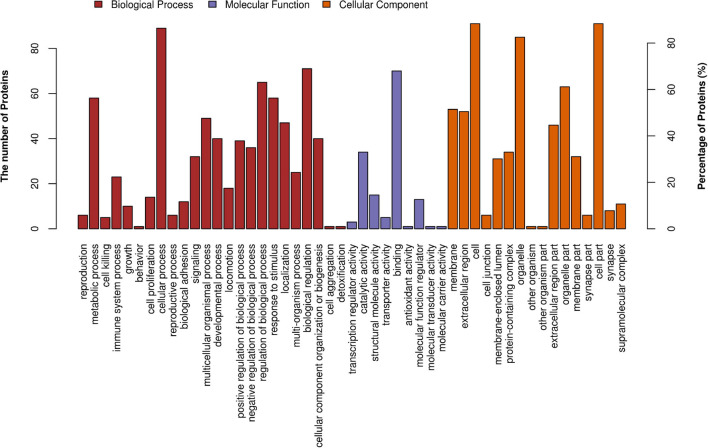
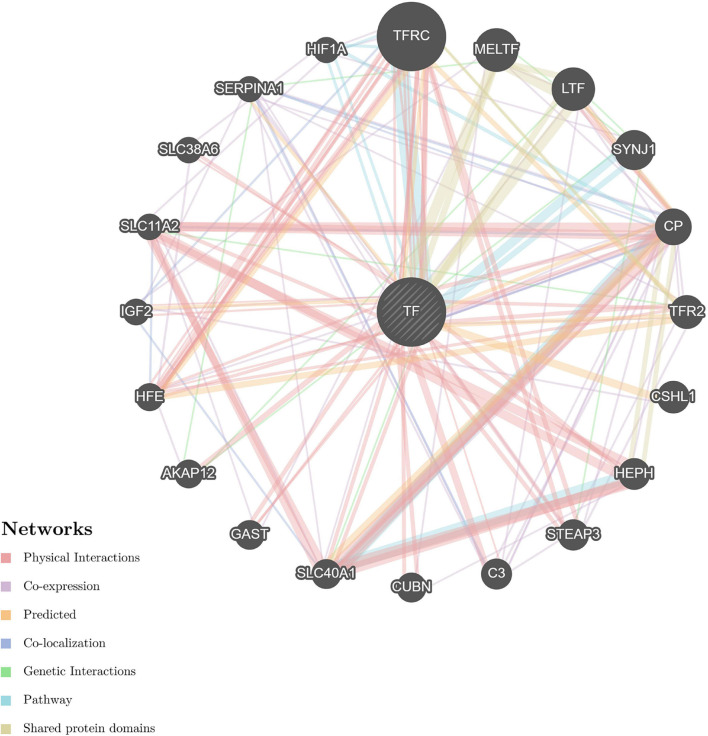
Myopia is a major public health concern with increasing global prevalence and is the leading cause of vision loss and complications. The potential role of the cornea, a substantial component of refractive power and the protective fortress of the eye, has been underestimated in the development of myopia. Our study acquired corneal stroma tissues from myopic patients undergoing femtosecond laser-assisted small incision lenticule extraction (SMILE) surgery and investigated the differential expression of circulating proteins between subjects with low and high myopia by means of high-throughput proteomic approaches-the quantitative tandem mass tag (TMT) labeling method and parallel reaction monitoring (PRM) validation. Across all corneal stroma tissue samples, a total of 2,455 proteins were identified qualitatively and quantitatively, 103 of which were differentially expressed between those with low and high myopia. The differentially abundant proteins (DAPs) between the groups of stroma samples mostly demonstrated catalytic activity and molecular function regulator and transporter activity and participated in metabolic processes, biological regulation, response to stimulus, and so forth. Pathway enrichment showed that mineral absorption, ferroptosis, and HIF-1 signaling pathways were activated in the human myopic cornea. Furthermore, TMT analysis and PRM validation revealed that the expression of ferritin light chain (FTL, P02792) and ferritin heavy chain (FTH1, P02794) was negatively associated with myopia development, while the expression of serotransferrin (TF, P02787) was positively related to myopia status. Overall, our results indicated that subjects with low and high myopia could have different proteomic profiles or signatures in the cornea. These findings revealed disturbances in iron metabolism and corneal oxidative stress in the more myopic eyes. Iron metabolic proteins could serve as an essential modulator in the pathogenesis of myopia.
Keywords: cornea; iron metabolism; myopia; oxidative stress; protein biomarkers; protein–protein interaction; signal.
Copyright © 2021 Chen, Wu, Wang, Zhai, Deng, Alzogool and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures








References
-
- Bostanci N., Selevsek N., Wolski W., Grossmann J., Bao K., Wahlander A., et al. (2018). Targeted proteomics guided by label-free quantitative proteome analysis in saliva reveal transition signatures from health to periodontal disease. Mol. Cell Proteomics 17 1392–1409. 10.1074/mcp.RA118.000718 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous