

Detection of Neanderthal Adaptively Introgressed Genetic Variants That Modulate Reporter Gene Expression in Human Immune Cells
- PMID: 34662402
- PMCID: PMC8760939
- DOI: 10.1093/molbev/msab304
Detection of Neanderthal Adaptively Introgressed Genetic Variants That Modulate Reporter Gene Expression in Human Immune Cells
Abstract
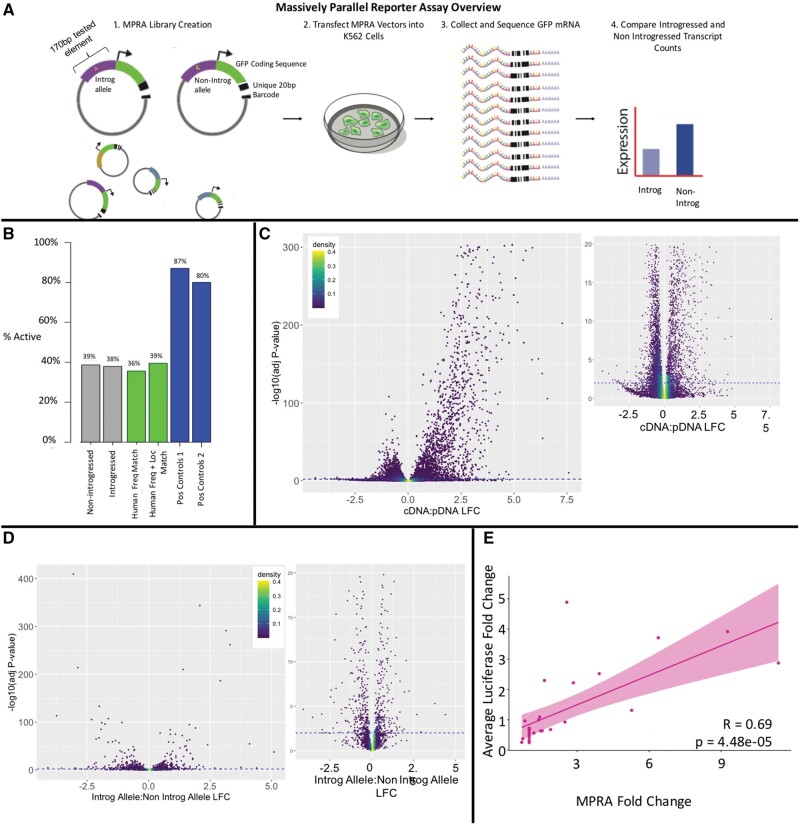
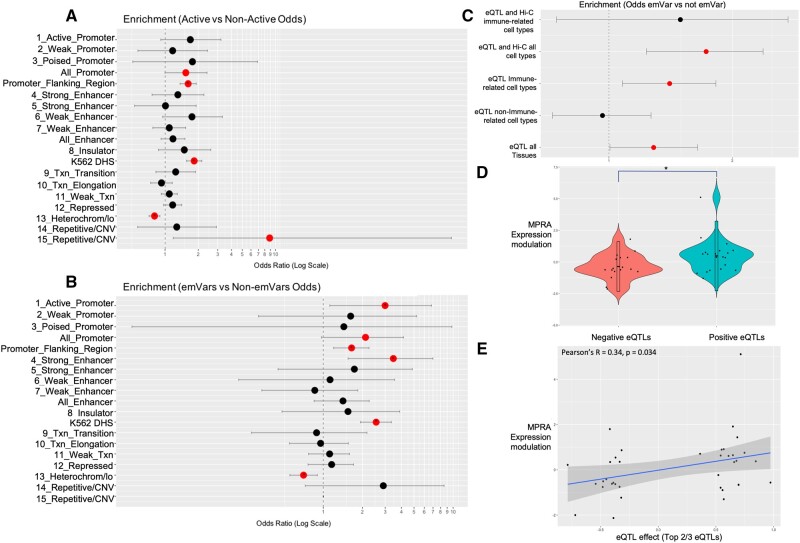
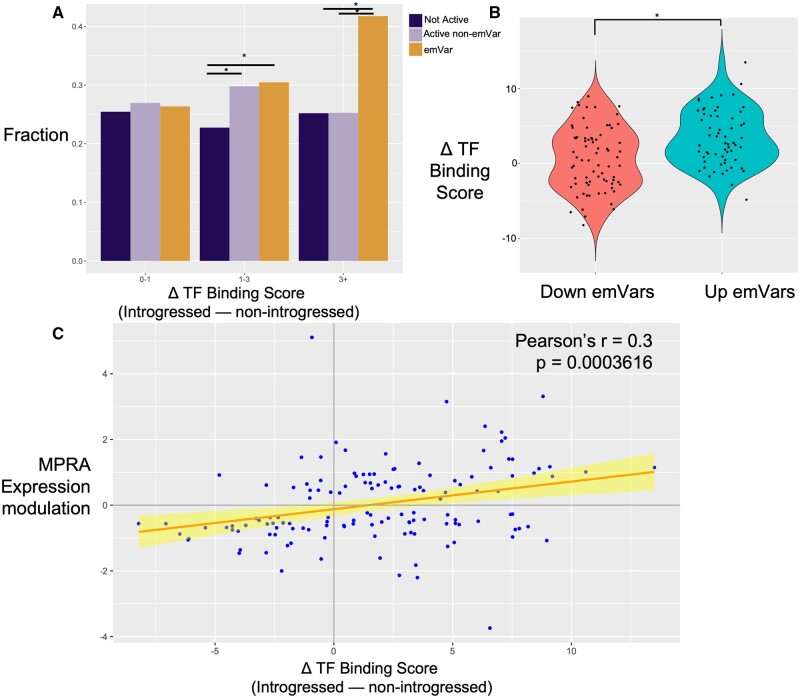
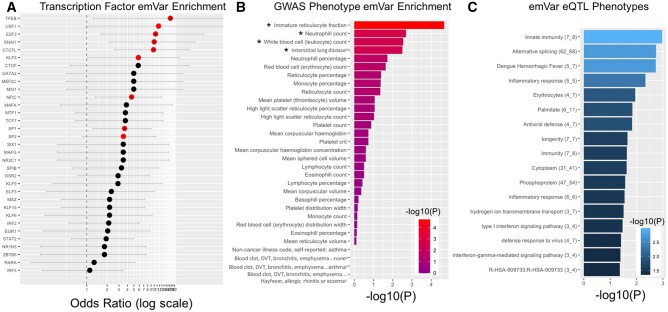
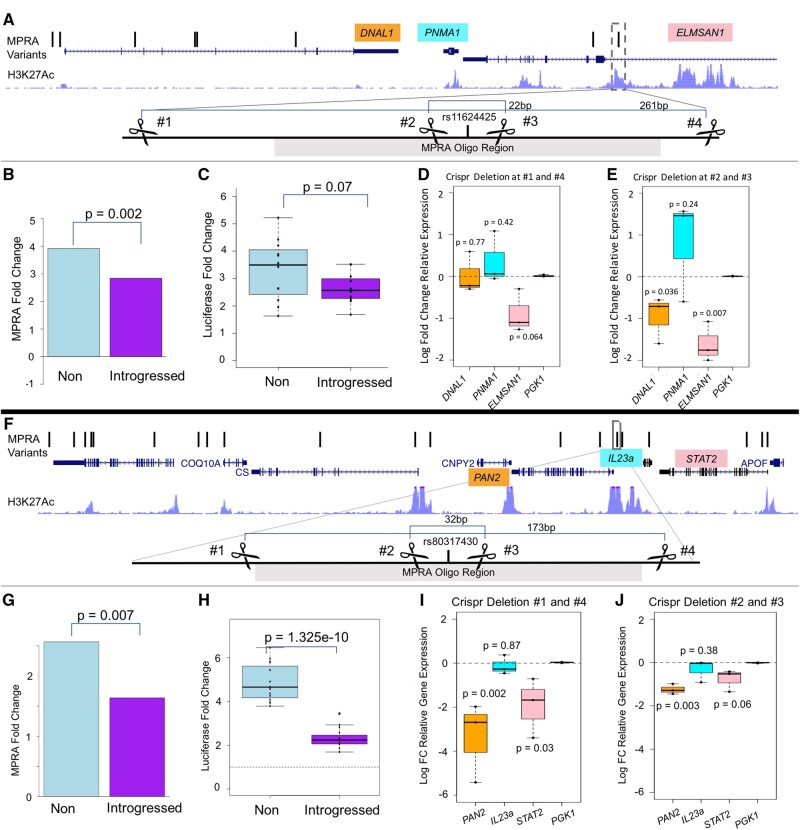
Although some variation introgressed from Neanderthals has undergone selective sweeps, little is known about its functional significance. We used a Massively Parallel Reporter Assay (MPRA) to assay 5,353 high-frequency introgressed variants for their ability to modulate the gene expression within 170 bp of endogenous sequence. We identified 2,548 variants in active putative cis-regulatory elements (CREs) and 292 expression-modulating variants (emVars). These emVars are predicted to alter the binding motifs of important immune transcription factors, are enriched for associations with neutrophil and white blood cell count, and are associated with the expression of genes that function in innate immune pathways including inflammatory response and antiviral defense. We combined the MPRA data with other data sets to identify strong candidates to be driver variants of positive selection including an emVar that may contribute to protection against severe COVID-19 response. We endogenously deleted two CREs containing expression-modulation variants linked to immune function, rs11624425 and rs80317430, identifying their primary genic targets as ELMSAN1, and PAN2 and STAT2, respectively, three genes differentially expressed during influenza infection. Overall, we present the first database of experimentally identified expression-modulating Neanderthal-introgressed alleles contributing to potential immune response in modern humans.
Keywords: adaptation; immune; introgression; massively parallel reporter assay; neandertal; positive selection.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Bath P, Algert C, Chapman N, Neal B, PROGRESS Collaborative Group. 2004. Association of mean platelet volume with risk of stroke among 3134 individuals with history of cerebrovascular disease. Stroke 35(3):622–626. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
