

Assessing Multiplex Tiling PCR Sequencing Approaches for Detecting Genomic Variants of SARS-CoV-2 in Municipal Wastewater
- PMID: 34665013
- PMCID: PMC8525555
- DOI: 10.1128/mSystems.01068-21
Assessing Multiplex Tiling PCR Sequencing Approaches for Detecting Genomic Variants of SARS-CoV-2 in Municipal Wastewater
Abstract
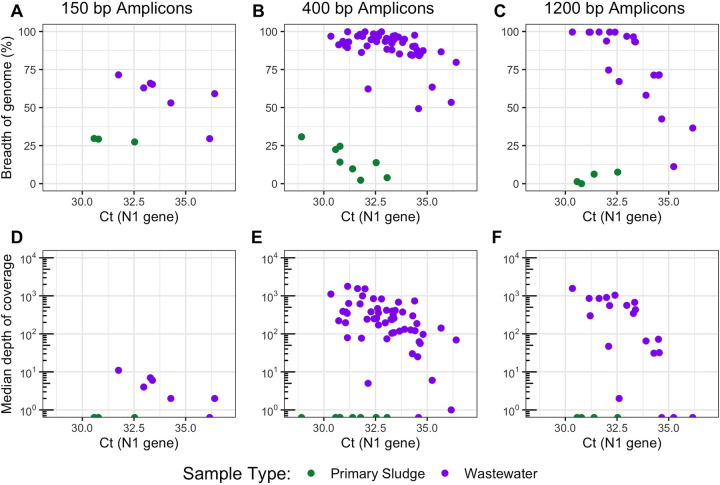
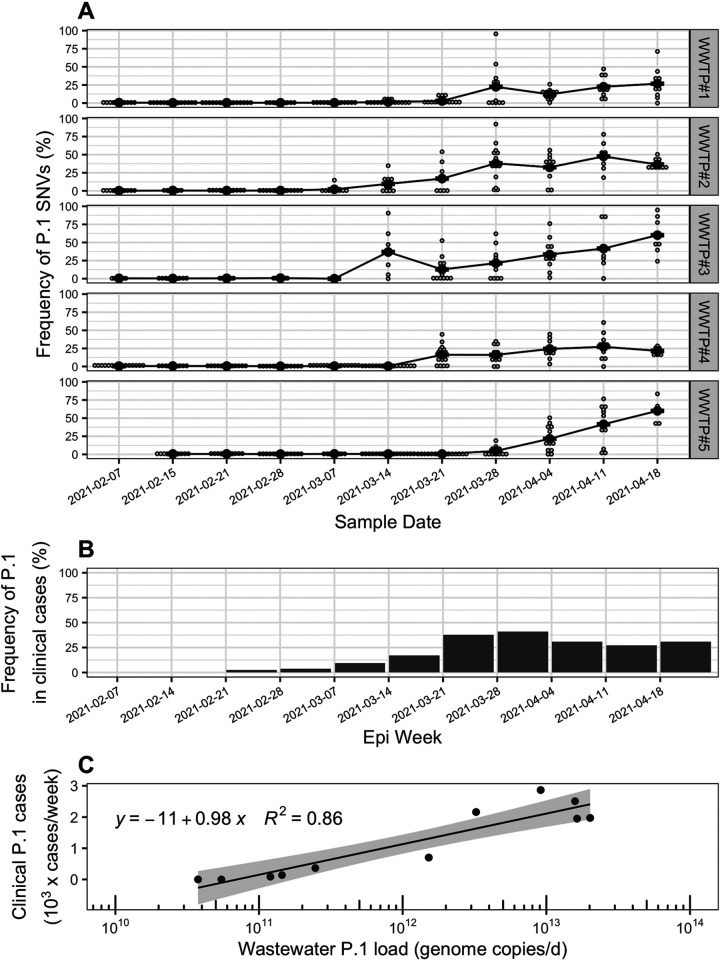
Wastewater-based genomic surveillance of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus shows promise to complement genomic epidemiology efforts. Multiplex tiling PCR is a desirable approach for targeted genome sequencing of SARS-CoV-2 in wastewater due to its low cost and rapid turnaround time. However, it is not clear how different multiplex tiling PCR primer schemes or wastewater sample matrices impact the resulting SARS-CoV-2 genome coverage. The objective of this work was to assess the performance of three different multiplex primer schemes, consisting of 150-bp, 400-bp, and 1,200-bp amplicons, as well as two wastewater sample matrices, influent wastewater and primary sludge, for targeted genome sequencing of SARS-CoV-2. Wastewater samples were collected weekly from five municipal wastewater treatment plants (WWTPs) in the Metro Vancouver region of British Columbia, Canada during a period of increased coronavirus disease 19 (COVID-19) case counts from February to April 2021. RNA extracted from clarified influent wastewater provided significantly higher genome coverage (breadth and median depth) than primary sludge samples across all primer schemes. Shorter amplicons appeared to be more resilient to sample RNA degradation but were hindered by greater primer pool complexity in the 150-bp scheme. The identified optimal primer scheme (400 bp) and sample matrix (influent) were capable of detecting the emergence of mutations associated with genomic variants of concern, for which the daily wastewater load significantly correlated with clinical case counts. Taken together, these results provide guidance on best practices for implementing wastewater-based genomic surveillance and demonstrate its ability to inform epidemiology efforts by detecting genomic variants of concern circulating within a geographic region. IMPORTANCE Monitoring the genomic characteristics of the SARS-CoV-2 virus circulating in a population can shed important insights into epidemiological aspects of the COVID-19 outbreak. Sequencing every clinical patient sample in a highly populous area is a difficult feat, and thus sequencing SARS-CoV-2 RNA in municipal wastewater offers great promise to augment genomic surveillance by characterizing a pooled population sample matrix, particularly during an escalating outbreak. Here, we assess different approaches and sample matrices for rapid targeted genome sequencing of SARS-CoV-2 in municipal wastewater. We demonstrate that the optimal approach is capable of detecting the emergence of SARS-CoV-2 genomic variants of concern, with strong correlations to clinical case data in the province of British Columbia. These results provide guidance on best practices on, as well as further support for, the application of wastewater genomic surveillance as a tool to augment current genomic epidemiology efforts.
Keywords: COVID-19; RNA; SARS-CoV-2; epidemiology; variants; wastewater; whole-genome sequencing.
Figures
Similar articles
-
RNA Viromics of Southern California Wastewater and Detection of SARS-CoV-2 Single-Nucleotide Variants.Appl Environ Microbiol. 2021 Nov 10;87(23):e0144821. doi: 10.1128/AEM.01448-21. Epub 2021 Sep 22. Appl Environ Microbiol. 2021. PMID: 34550753 Free PMC article.
-
Emergency SARS-CoV-2 Variants of Concern: Novel Multiplex Real-Time RT-PCR Assay for Rapid Detection and Surveillance.Microbiol Spectr. 2022 Feb 23;10(1):e0251321. doi: 10.1128/spectrum.02513-21. Epub 2022 Feb 23. Microbiol Spectr. 2022. PMID: 35196812 Free PMC article.
-
Wastewater genomic sequencing for SARS-CoV-2 variants surveillance in wastewater-based epidemiology applications.Water Res. 2023 Oct 1;244:120444. doi: 10.1016/j.watres.2023.120444. Epub 2023 Aug 3. Water Res. 2023. PMID: 37579567
-
A review on presence, survival, disinfection/removal methods of coronavirus in wastewater and progress of wastewater-based epidemiology.J Environ Chem Eng. 2020 Oct;8(5):104317. doi: 10.1016/j.jece.2020.104317. Epub 2020 Aug 5. J Environ Chem Eng. 2020. PMID: 32834991 Free PMC article. Review.
-
An interpretative review of the wastewater-based surveillance of the SARS-CoV-2: where do we stand on its presence and concern?Front Microbiol. 2024 Jan 22;15:1338100. doi: 10.3389/fmicb.2024.1338100. eCollection 2024. Front Microbiol. 2024. PMID: 38318336 Free PMC article. Review.
Cited by
-
Reconstructing SARS-CoV-2 lineages from mixed wastewater sequencing data.Sci Rep. 2024 Aug 31;14(1):20273. doi: 10.1038/s41598-024-70416-4. Sci Rep. 2024. PMID: 39217200 Free PMC article.
-
Early detection and surveillance of SARS-CoV-2 genomic variants in wastewater using COJAC.Nat Microbiol. 2022 Aug;7(8):1151-1160. doi: 10.1038/s41564-022-01185-x. Epub 2022 Jul 18. Nat Microbiol. 2022. PMID: 35851854 Free PMC article.
-
SARS-CoV-2 Monitoring in Wastewater Reveals Novel Variants and Biomarkers of Infection.Viruses. 2022 Sep 13;14(9):2032. doi: 10.3390/v14092032. Viruses. 2022. PMID: 36146835 Free PMC article.
-
Development of Primer Panels for Whole-Genome Amplification and Sequencing of Human Seasonal Coronaviruses: hCoV-OC43, hCoV-HKU1, hCoV-229E, and hCoV-NL63.Viruses. 2024 Dec 25;17(1):13. doi: 10.3390/v17010013. Viruses. 2024. PMID: 39861802 Free PMC article.
-
Emerging Multiplex Nucleic Acid Diagnostic Tests for Combating COVID-19.Biosensors (Basel). 2022 Nov 7;12(11):978. doi: 10.3390/bios12110978. Biosensors (Basel). 2022. PMID: 36354487 Free PMC article. Review.
References
-
- Bedford T, Greninger AL, Roychoudhury P, Starita LM, Famulare M, Huang M-L, Nalla A, Pepper G, Reinhardt A, Xie H, Shrestha L, Nguyen TN, Adler A, Brandstetter E, Cho S, Giroux D, Han PD, Fay K, Frazar CD, Ilcisin M, Lacombe K, Lee J, Kiavand A, Richardson M, Sibley TR, Truong M, Wolf CR, Nickerson DA, Rieder MJ, Englund JA, Hadfield J, Hodcroft EB, Huddleston J, Moncla LH, Müller NF, Neher RA, Deng X, Gu W, Federman S, Chiu C, Duchin JS, Gautom R, Melly G, Hiatt B, Dykema P, Lindquist S, Queen K, Tao Y, Uehara A, Tong S, Seattle Flu Study Investigators, et al. . 2020. Cryptic transmission of SARS-CoV-2 in Washington state. Science 370:571–575. doi:10.1126/science.abc0523. - DOI - PMC - PubMed
-
- Candido DS, Claro IM, de Jesus JG, Souza WM, Moreira FRR, Dellicour S, Mellan TA, du Plessis L, Pereira RHM, Sales FCS, Manuli ER, Thézé J, Almeida L, Menezes MT, Voloch CM, Fumagalli MJ, Coletti TM, da Silva CAM, Ramundo MS, Amorim MR, Hoeltgebaum HH, Mishra S, Gill MS, Carvalho LM, Buss LF, Prete CA, Ashworth J, Nakaya HI, Peixoto PS, Brady OJ, Nicholls SM, Tanuri A, Rossi ÁD, Braga CKV, Gerber AL, de Guimarães APC, Gaburo N, Alencar CS, Ferreira ACS, Lima CX, Levi JE, Granato C, Ferreira GM, Francisco RS, Granja F, Garcia MT, Moretti ML, Perroud MW, Castiñeiras TMPP, Lazari CS, Hill SC, de Santos AAS, Simeoni CL, Forato J, Sposito AC, Schreiber AZ, Santos MNN, Brazil-UK Centre for Arbovirus Discovery, Diagnosis, Genomics and Epidemiology (CADDE) Genomic Network, et al. . 2020. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 369:1255–1260. doi:10.1126/science.abd2161. - DOI - PMC - PubMed
-
- Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday JD, Pearson CAB, Russell TW, Tully DC, Washburne AD, Wenseleers T, Gimma A, Waites W, Wong KLM, van Zandvoort K, Silverman JD, Diaz-Ordaz K, Keogh R, Eggo RM, Funk S, Jit M, Atkins KE, Edmunds WJ, CMMID COVID-19 Working Group, COVID-19 Genomics UK (COG-UK) Consortium . 2021. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 372:eabg3055. doi:10.1126/science.abg3055. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
