

Genetic testing for familial hypercholesterolemia-past, present, and future
- PMID: 34666015
- PMCID: PMC8572866
- DOI: 10.1016/j.jlr.2021.100139
Genetic testing for familial hypercholesterolemia-past, present, and future
Abstract
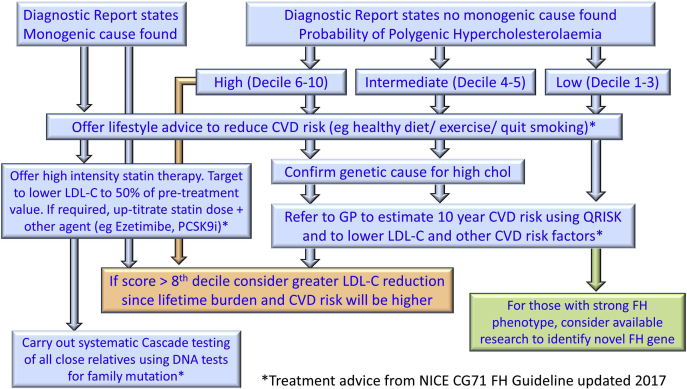
In the early 1980s, the Nobel Prize winning cellular and molecular work of Mike Brown and Joe Goldstein led to the identification of the LDL receptor gene as the first gene where mutations cause the familial hypercholesterolemia (FH) phenotype. We now know that autosomal dominant monogenic FH can be caused by pathogenic variants of three additional genes (APOB/PCSK9/APOE) and that the plasma LDL-C concentration and risk of premature coronary heart disease differs according to the specific locus and associated molecular cause. It is now possible to use next-generation sequencing to sequence all exons of all four genes, processing 96 patient samples in one sequencing run, increasing the speed of test results, and reducing costs. This has resulted in the identification of not only many novel FH-causing variants but also some variants of unknown significance, which require further evidence to classify as pathogenic or benign. The identification of the FH-causing variant in an index case can be used as an unambiguous and rapid test for other family members. An FH-causing variant can be found in 20-40% of patients with the FH phenotype, and we now appreciate that in the majority of patients without a monogenic cause, a polygenic etiology for their phenotype is highly likely. Compared with those with a monogenic cause, these patients have significantly lower risk of future coronary heart disease. The use of these molecular genetic diagnostic methods in the characterization of FH is a prime example of the utility of precision or personalized medicine.
Keywords: LDL-C; LDLR; SNP score; clinical utility; coronary heart disease; index case; monogenic; next-generation sequencing; polygenic; variants of unknown significance.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest M. F. reports speakers' fees from Sanofi, and S. E. H. reports receiving fees for Advisory Boards of Novartis and Amryt. S. E. H. is the Medical Director of a University College London spin-out company StoreGene that offers to clinicians genetic testing for patients with FH. S. E. H. directs the UK Children's FH Register, which has been supported by a grant from Pfizer (grant no.: 24052829) given by the International Atherosclerosis Society. M. W. reported speaker fees from Amgen and Akcea. A. T.-B. declares no conflict of interest with the contents of this article.
Figures



References
-
- Boren J., Ekstrom U., Agren B., Nilsson-Ehle P., Innerarity T.L. The molecular mechanism for the genetic disorder familial defective apolipoprotein B100. J. Biol. Chem. 2001;276:9214–9218. - PubMed
-
- Abifadel M., Varret M., Rabes J.P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derre A., Villeger L., Farnier M., Beucler I., Bruckert E. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003;34:154–156. - PubMed
-
- Marduel M., Ouguerram K., Serre V., Bonnefont-Rousselot D., Marques-Pinheiro A., Erik Berge K., Devillers M., Luc G., Lecerf J.M., Tosolini L., Erlich D., Peloso G.M., Stitziel N., Nitchke P., Jais J.P. Description of a large family with autosomal dominant hypercholesterolemia associated with the APOE p.Leu167del mutation. Hum. Mutat. 2013;34:83–87. - PMC - PubMed
-
- Iacocca M.A., Chora J.R., Carrie A., Freiberger T., Leigh S.E., Defesche J.C., Kurtz C.L., DiStefano M.T., Santos R.D., Humphries S.E., Mata P., Jannes C.E., Hooper A.J., Wilemon K.A., Benlian P. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum. Mutat. 2018;39:1631–1640. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
