

Interpretable machine learning for genomics
- PMID: 34669035
- PMCID: PMC8527313
- DOI: 10.1007/s00439-021-02387-9
Interpretable machine learning for genomics
Abstract
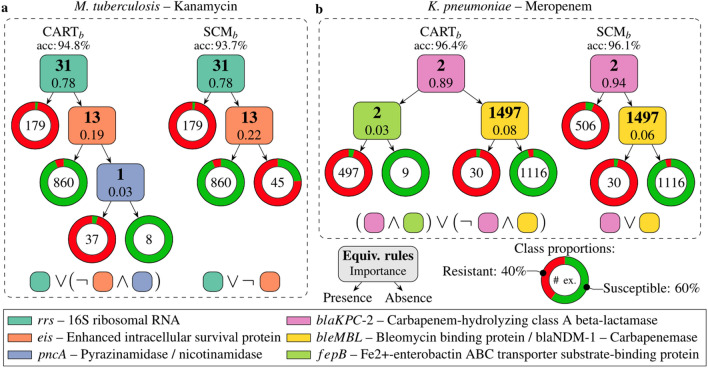
High-throughput technologies such as next-generation sequencing allow biologists to observe cell function with unprecedented resolution, but the resulting datasets are too large and complicated for humans to understand without the aid of advanced statistical methods. Machine learning (ML) algorithms, which are designed to automatically find patterns in data, are well suited to this task. Yet these models are often so complex as to be opaque, leaving researchers with few clues about underlying mechanisms. Interpretable machine learning (iML) is a burgeoning subdiscipline of computational statistics devoted to making the predictions of ML models more intelligible to end users. This article is a gentle and critical introduction to iML, with an emphasis on genomic applications. I define relevant concepts, motivate leading methodologies, and provide a simple typology of existing approaches. I survey recent examples of iML in genomics, demonstrating how such techniques are increasingly integrated into research workflows. I argue that iML solutions are required to realize the promise of precision medicine. However, several open challenges remain. I examine the limitations of current state-of-the-art tools and propose a number of directions for future research. While the horizon for iML in genomics is wide and bright, continued progress requires close collaboration across disciplines.
© 2021. The Author(s).
Conflict of interest statement
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Figures
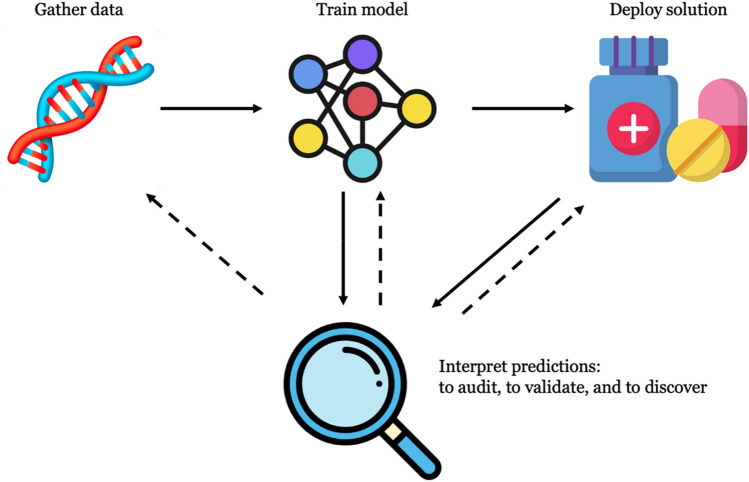
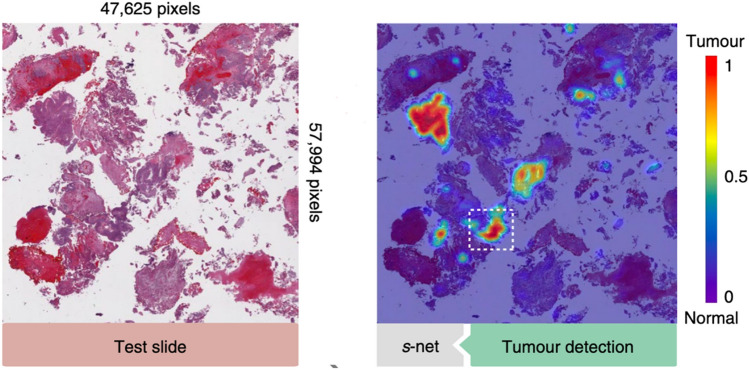
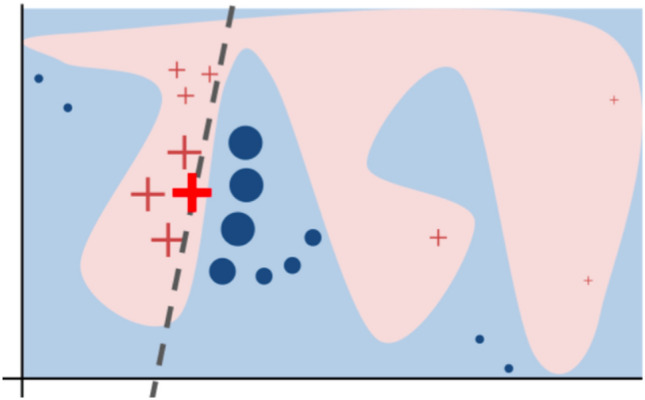
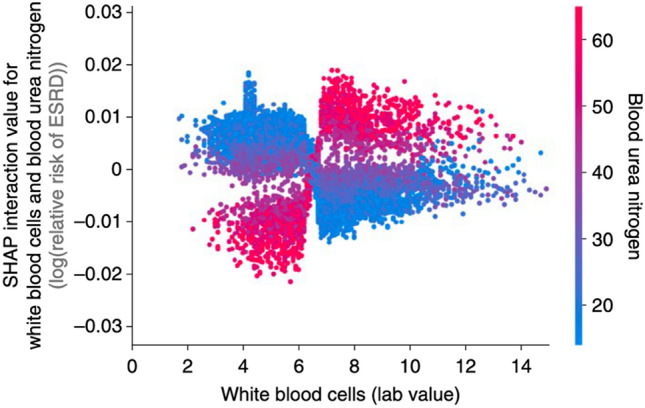
References
-
- Aas K, Jullum M, Løland A. Explaining individual predictions when features are dependent: more accurate approximations to Shapley values. Artif Intell. 2021;298:103502. doi: 10.1016/j.artint.2021.103502. - DOI
-
- Adadi A, Berrada M. Peeking inside the black-box: a survey on explainable artificial intelligence (XAI) IEEE Access. 2018;6:52138–52160. doi: 10.1109/ACCESS.2018.2870052. - DOI
-
- Angelino E, Larus-Stone N, Alabi D, Seltzer M, Rudin C. Learning certifiably optimal rule lists for categorical data. J Mach Learn Res. 2018;18(234):1–78.
-
- Anguita-Ruiz A, Segura-Delgado A, Alcalá R, Aguilera CM, Alcalá-Fdez J. eXplainable artificial intelligence (XAI) for the identification of biologically relevant gene expression patterns in longitudinal human studies, insights from obesity research. PLoS Comput Biol. 2020;16(4):e1007792. doi: 10.1371/journal.pcbi.1007792. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
