

Bayesian log-normal deconvolution for enhanced in silico microdissection of bulk gene expression data
- PMID: 34671028
- PMCID: PMC8528834
- DOI: 10.1038/s41467-021-26328-2
Bayesian log-normal deconvolution for enhanced in silico microdissection of bulk gene expression data
Abstract
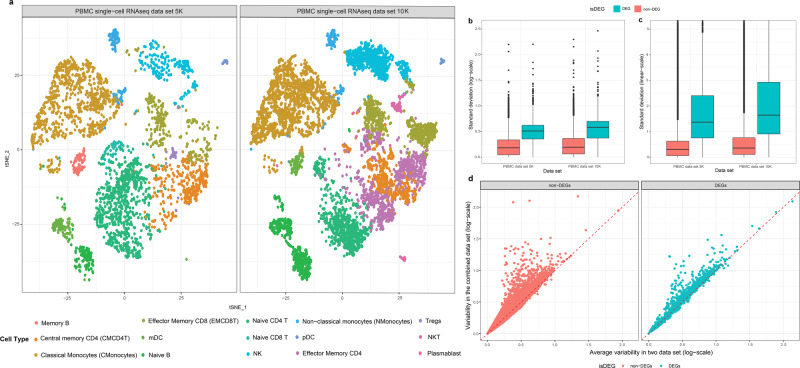
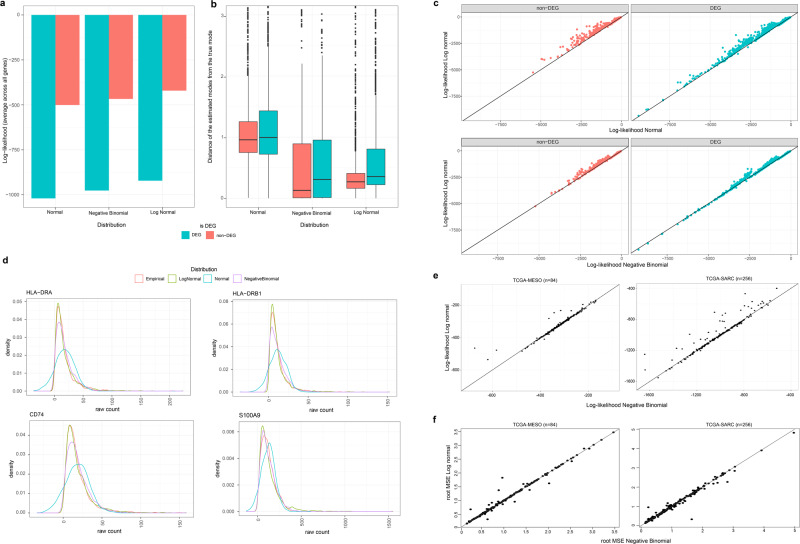
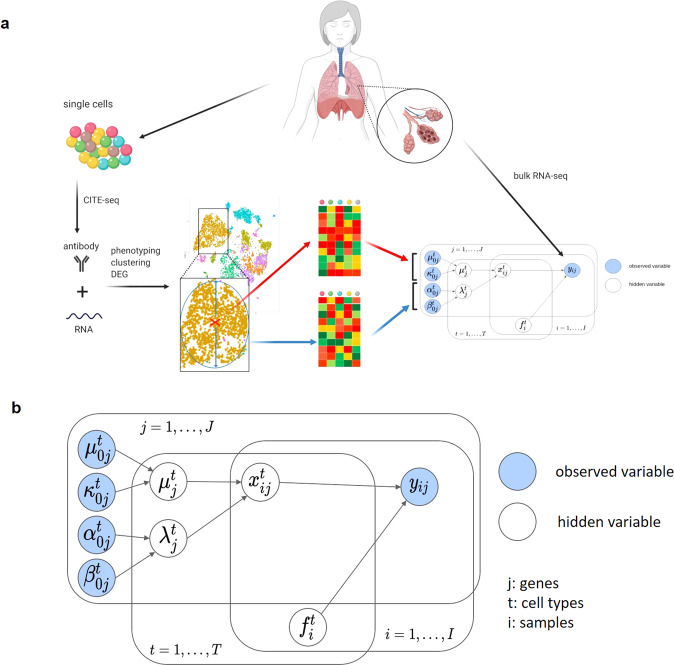
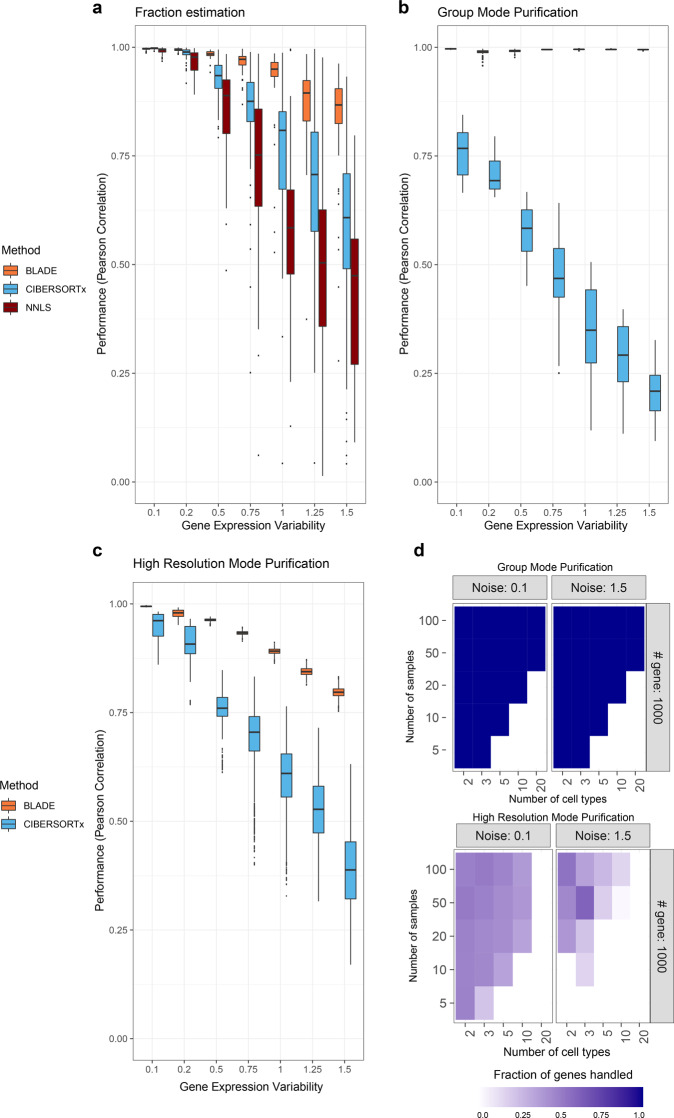
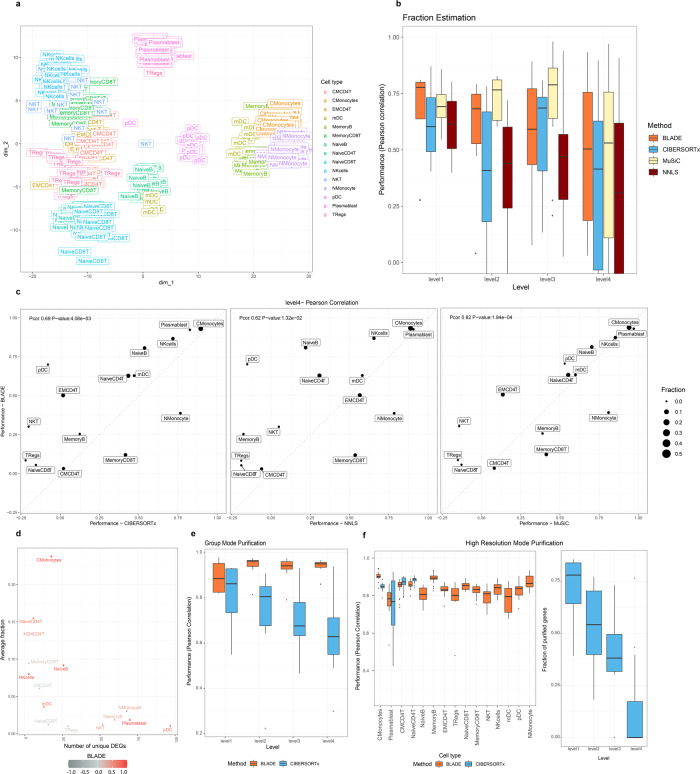
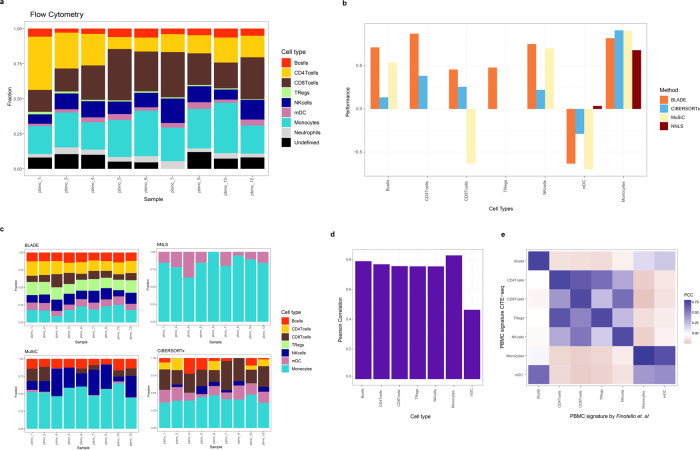
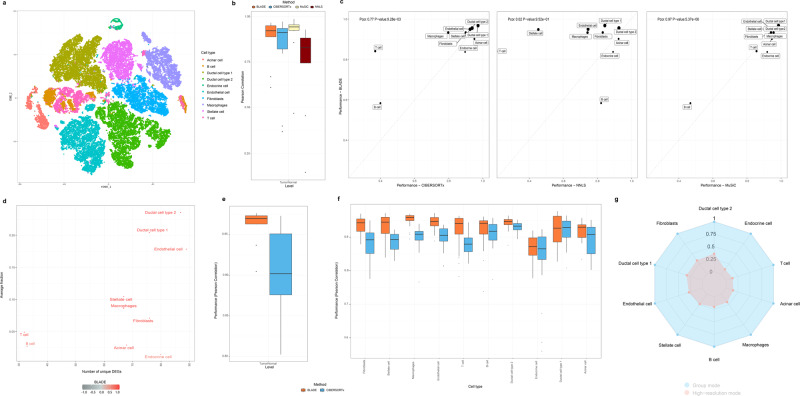
Deconvolution of bulk gene expression profiles into the cellular components is pivotal to portraying tissue's complex cellular make-up, such as the tumor microenvironment. However, the inherently variable nature of gene expression requires a comprehensive statistical model and reliable prior knowledge of individual cell types that can be obtained from single-cell RNA sequencing. We introduce BLADE (Bayesian Log-normAl Deconvolution), a unified Bayesian framework to estimate both cellular composition and gene expression profiles for each cell type. Unlike previous comprehensive statistical approaches, BLADE can handle > 20 types of cells due to the efficient variational inference. Throughout an intensive evaluation with > 700 simulated and real datasets, BLADE demonstrated enhanced robustness against gene expression variability and better completeness than conventional methods, in particular, to reconstruct gene expression profiles of each cell type. In summary, BLADE is a powerful tool to unravel heterogeneous cellular activity in complex biological systems from standard bulk gene expression data.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
