

The type I interferonopathies: 10 years on
- PMID: 34671122
- PMCID: PMC8527296
- DOI: 10.1038/s41577-021-00633-9
The type I interferonopathies: 10 years on
Abstract
As brutally demonstrated by the COVID-19 pandemic, an effective immune system is essential for survival. Developed over evolutionary time, viral nucleic acid detection is a central pillar in the defensive armamentarium used to combat foreign microbial invasion. To ensure cellular homeostasis, such a strategy necessitates the efficient discrimination of pathogen-derived DNA and RNA from that of the host. In 2011, it was suggested that an upregulation of type I interferon signalling might serve as a defining feature of a novel set of Mendelian inborn errors of immunity, where antiviral sensors are triggered by host nucleic acids due to a failure of self versus non-self discrimination. These rare disorders have played a surprisingly significant role in informing our understanding of innate immunity and the relevance of type I interferon signalling for human health and disease. Here we consider what we have learned in this time, and how the field may develop in the future.
© 2021. Springer Nature Limited.
Conflict of interest statement
The authors declare working as consultants for Related Sciences.
Figures
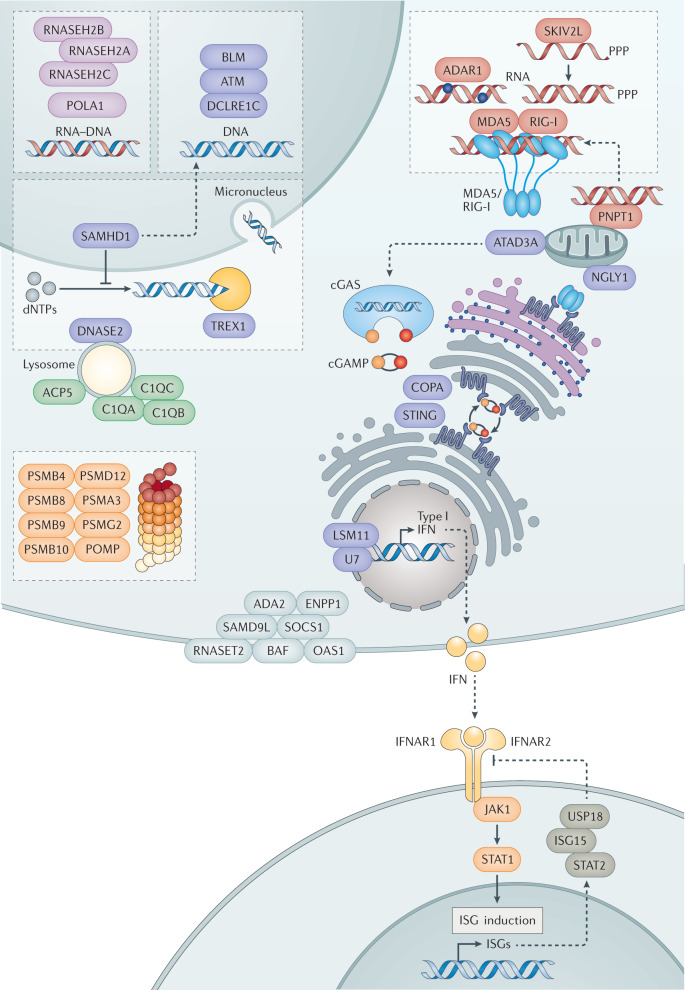
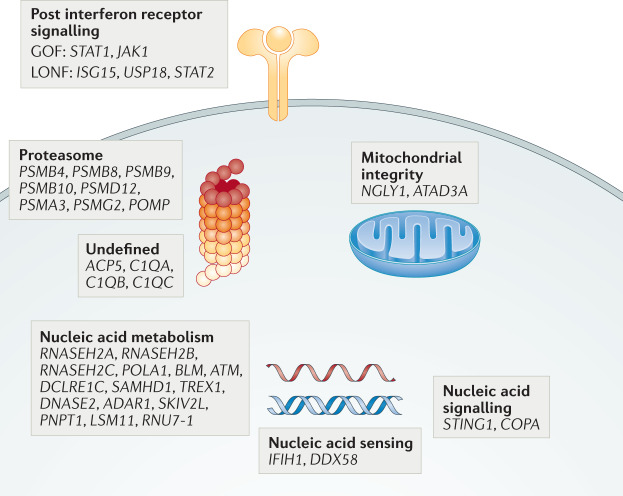
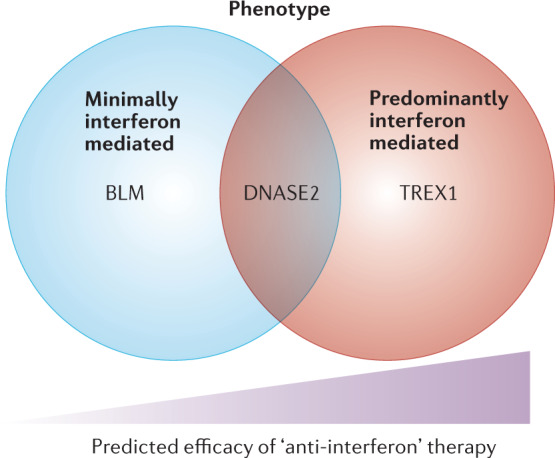
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
