

Proximal-end bias from in-vitro reconstituted nucleosomes and the result on downstream data analysis
- PMID: 34673804
- PMCID: PMC8530345
- DOI: 10.1371/journal.pone.0258737
Proximal-end bias from in-vitro reconstituted nucleosomes and the result on downstream data analysis
Abstract
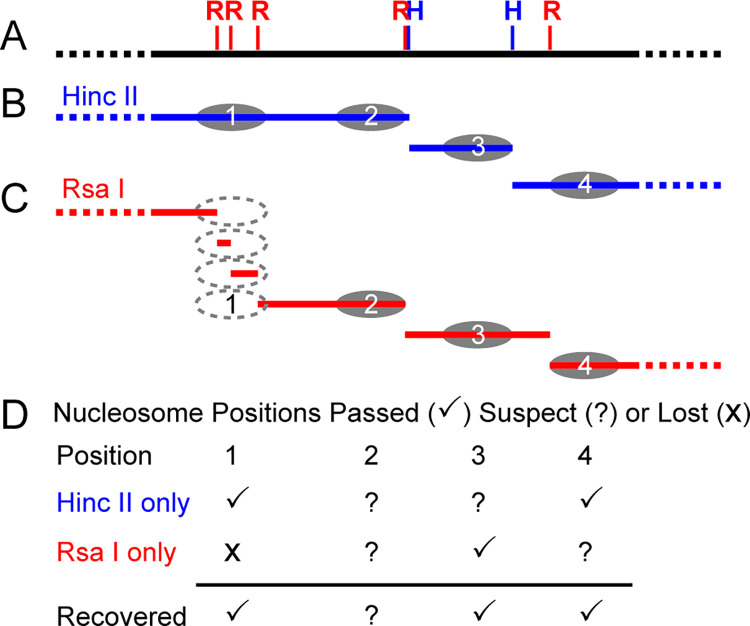
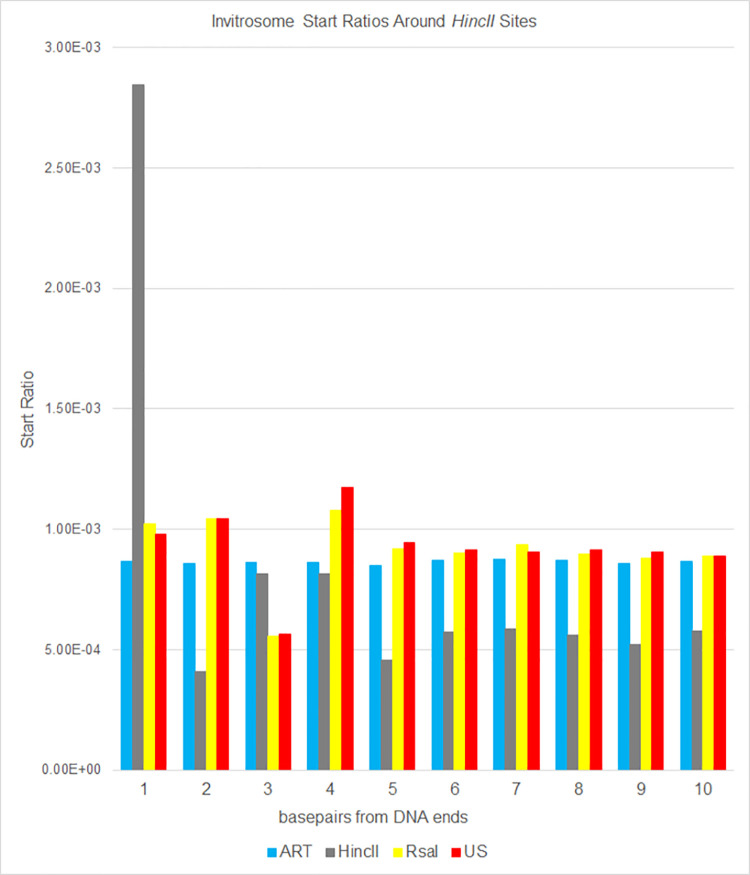
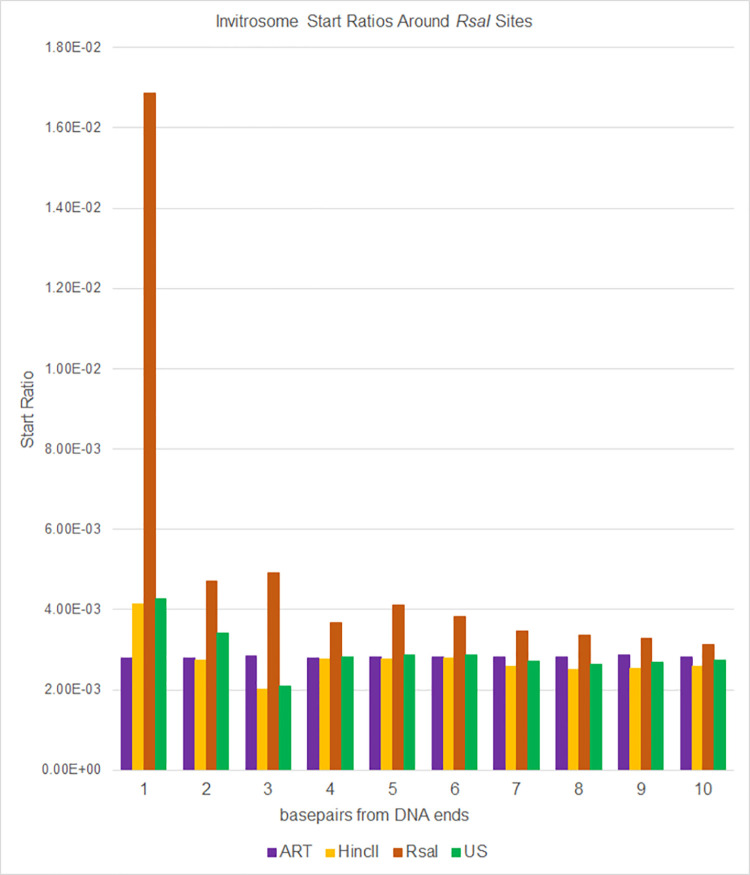
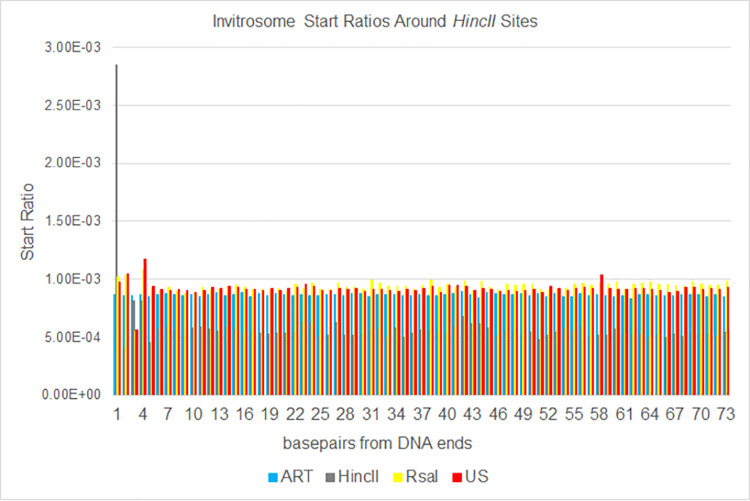
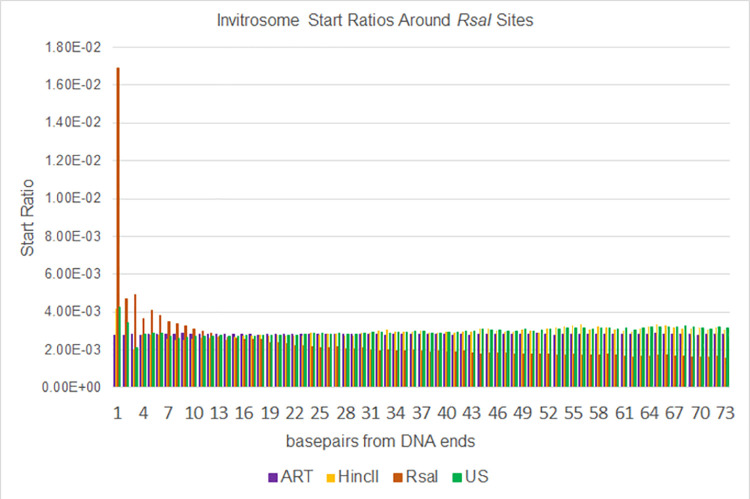
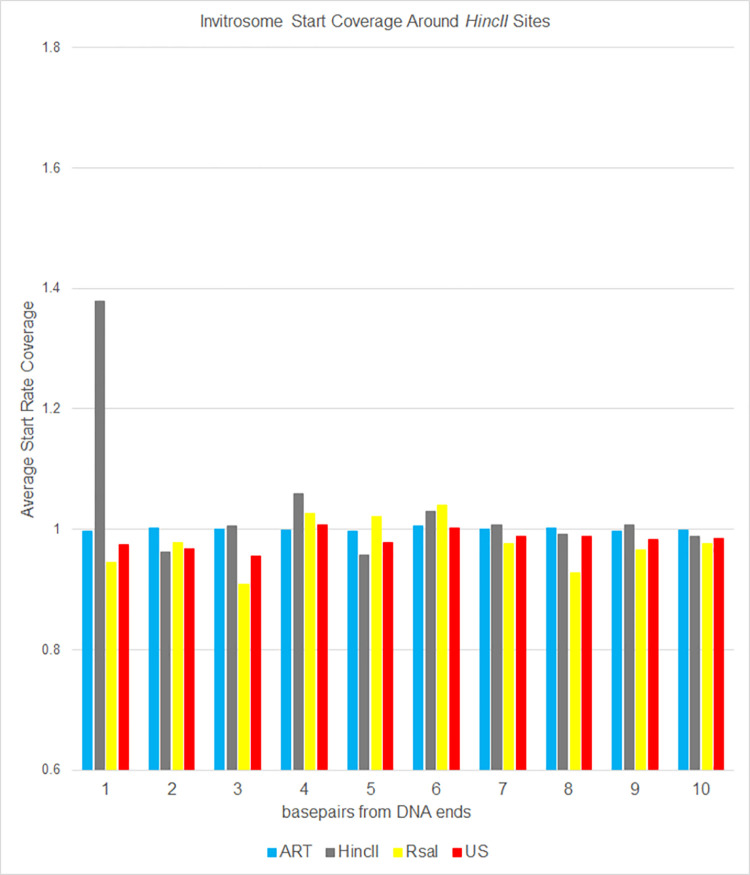
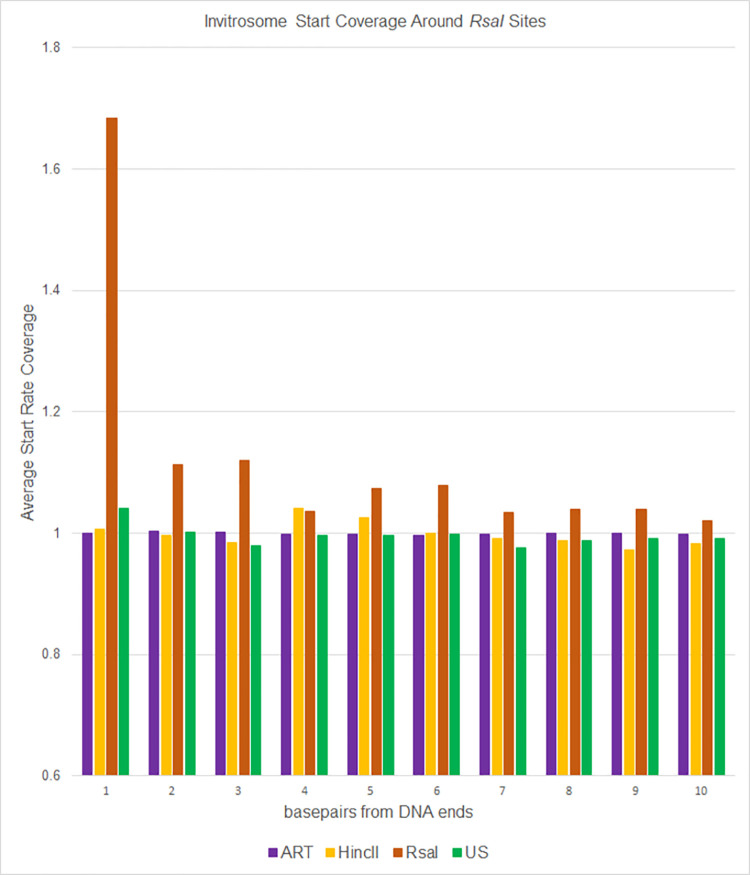
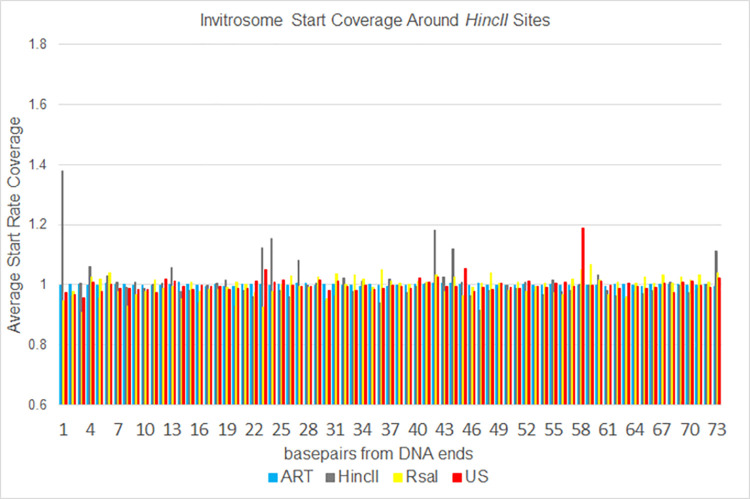
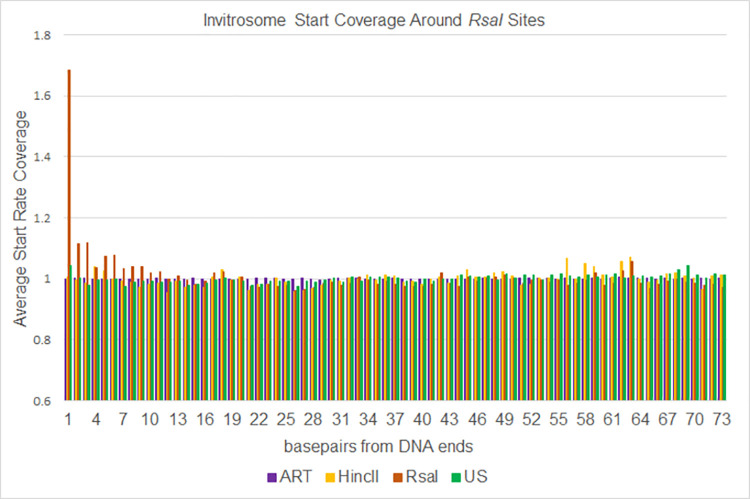
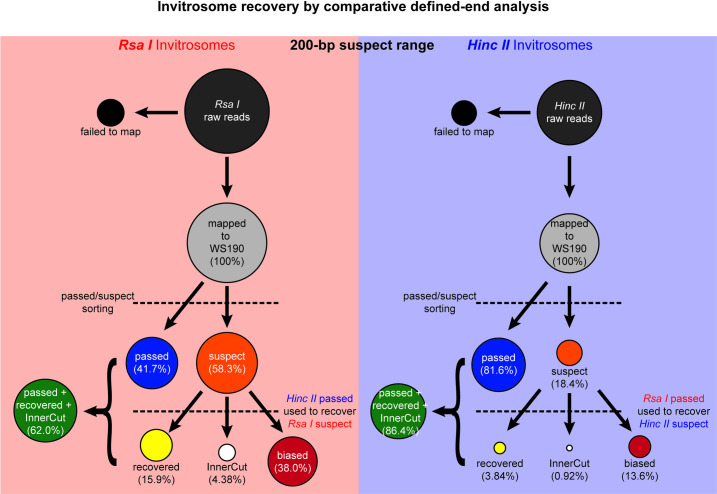
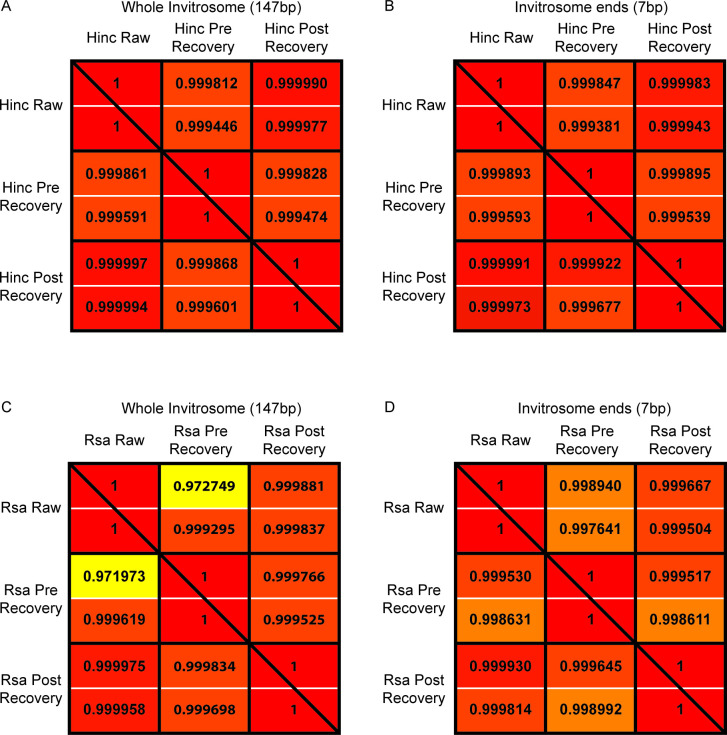
The most basic level of eukaryotic gene regulation is the presence or absence of nucleosomes on DNA regulatory elements. In an effort to elucidate in vivo nucleosome patterns, in vitro studies are frequently used. In vitro, short DNA fragments are more favorable for nucleosome formation, increasing the likelihood of nucleosome occupancy. This may in part result from the fact that nucleosomes prefer to form on the terminal ends of linear DNA. This phenomenon has the potential to bias in vitro reconstituted nucleosomes and skew results. If the ends of DNA fragments are known, the reads falling close to the ends are typically discarded. In this study we confirm the phenomenon of end bias of in vitro nucleosomes. We describe a method in which nearly identical libraries, with different known ends, are used to recover nucleosomes which form towards the terminal ends of fragmented DNA. Finally, we illustrate that although nucleosomes prefer to form on DNA ends, it does not appear to skew results or the interpretation thereof.
Conflict of interest statement
Two authors, DAB and CEB, are brothers. CEB owns and operates the software consulting company, Qubit Software LLC. The contributions of Qubit Software LLC were strictly pro bono, and the custom codes written are publicly available. This does not alter our adherence to PLOS ONE policies on sharing data and materials. Thus the authors declare no conflict of interest.
Figures

Similar articles
-
Global remodeling of nucleosome positions in C. elegans.BMC Genomics. 2013 Apr 26;14:284. doi: 10.1186/1471-2164-14-284. BMC Genomics. 2013. PMID: 23622142 Free PMC article.
-
Nucleosome fragility is associated with future transcriptional response to developmental cues and stress in C. elegans.Genome Res. 2017 Jan;27(1):75-86. doi: 10.1101/gr.208173.116. Epub 2016 Nov 14. Genome Res. 2017. PMID: 27979995 Free PMC article.
-
Visible periodicity of strong nucleosome DNA sequences.J Biomol Struct Dyn. 2015;33(1):1-9. doi: 10.1080/07391102.2013.855143. Epub 2013 Nov 22. J Biomol Struct Dyn. 2015. PMID: 24266748
-
Nucleosome positioning and spacing: from genome-wide maps to single arrays.Essays Biochem. 2019 Apr 23;63(1):5-14. doi: 10.1042/EBC20180058. Print 2019 Apr 23. Essays Biochem. 2019. PMID: 31015380 Review.
-
Nucleosome positioning in yeasts: methods, maps, and mechanisms.Chromosoma. 2015 Jun;124(2):131-51. doi: 10.1007/s00412-014-0501-x. Epub 2014 Dec 23. Chromosoma. 2015. PMID: 25529773 Review.
Cited by
-
Manipulating chromatin architecture in C. elegans.Epigenetics Chromatin. 2022 Nov 29;15(1):38. doi: 10.1186/s13072-022-00472-5. Epigenetics Chromatin. 2022. PMID: 36443798 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
