

SUFU haploinsufficiency causes a recognisable neurodevelopmental phenotype at the mild end of the Joubert syndrome spectrum
- PMID: 34675124
- PMCID: PMC9411896
- DOI: 10.1136/jmedgenet-2021-108114
SUFU haploinsufficiency causes a recognisable neurodevelopmental phenotype at the mild end of the Joubert syndrome spectrum
Abstract
Background: Joubert syndrome (JS) is a recessively inherited ciliopathy characterised by congenital ocular motor apraxia (COMA), developmental delay (DD), intellectual disability, ataxia, multiorgan involvement, and a unique cerebellar and brainstem malformation. Over 40 JS-associated genes are known with a diagnostic yield of 60%-75%.In 2018, we reported homozygous hypomorphic missense variants of the SUFU gene in two families with mild JS. Recently, heterozygous truncating SUFU variants were identified in families with dominantly inherited COMA, occasionally associated with mild DD and subtle cerebellar anomalies.
Methods: We reanalysed next generation sequencing (NGS) data in two cohorts comprising 1097 probands referred for genetic testing of JS genes.
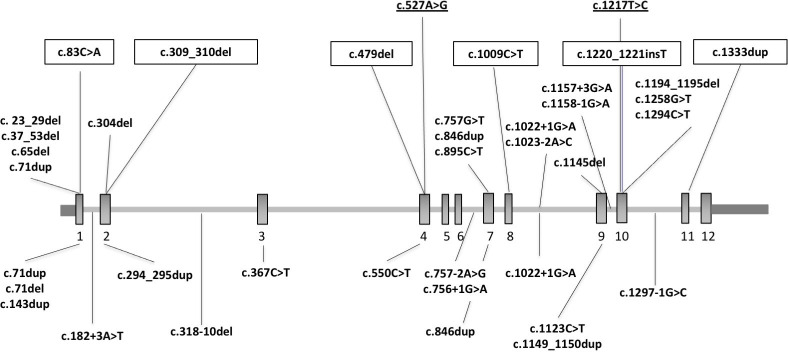
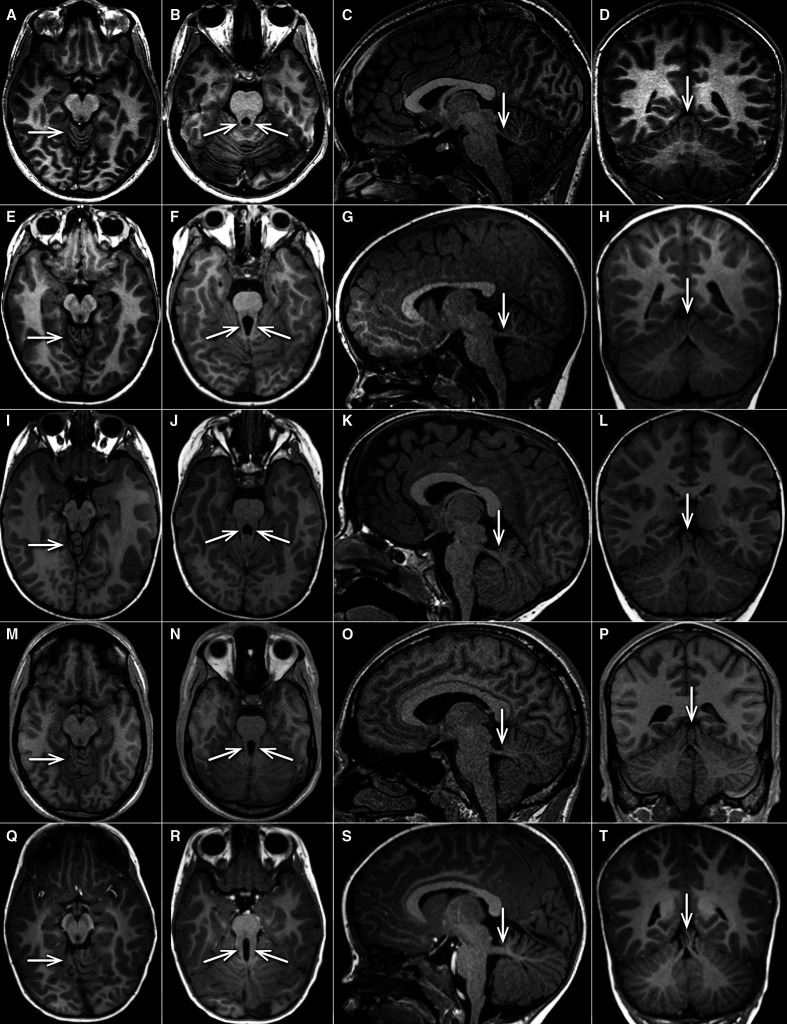
Results: Heterozygous truncating and splice-site SUFU variants were detected in 22 patients from 17 families (1.5%) with strong male prevalence (86%), and in 8 asymptomatic parents. Patients presented with COMA, hypotonia, ataxia and mild DD, and only a third manifested intellectual disability of variable severity. Brain MRI showed consistent findings characterised by vermis hypoplasia, superior cerebellar dysplasia and subtle-to-mild abnormalities of the superior cerebellar peduncles. The same pattern was observed in two out of three tested asymptomatic parents.
Conclusion: Heterozygous truncating or splice-site SUFU variants cause a novel neurodevelopmental syndrome encompassing COMA and mild JS, which likely represent overlapping entities. Variants can arise de novo or be inherited from a healthy parent, representing the first cause of JS with dominant inheritance and reduced penetrance. Awareness of this condition will increase the diagnostic yield of JS genetic testing, and allow appropriate counselling about prognosis, medical monitoring and recurrence risk.
Keywords: and neonatal diseases and abnormalities; central nervous system diseases; cerebellar diseases; congenital; early diagnosis; genetic variation; hereditary.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Nuovo S, Bacigalupo I, Ginevrino M, Battini R, Bertini E, Borgatti R, Casella A, Micalizzi A, Nardella M, Romaniello R, Serpieri V, Zanni G, Valente EM, Vanacore N, JS Italian Study Group . Age and sex prevalence estimate of Joubert syndrome in Italy. Neurology 2020;94:e797–801. 10.1212/WNL.0000000000008996 - DOI - PMC - PubMed
-
- Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, Chen ML, D'Arrigo S, Glass IA, Heller T, Héon E, Hildebrandt F, Joshi N, Knutzen D, Kroes HY, Mack SH, Nuovo S, Parisi MA, Snow J, Summers AC, Symons JM, Zein WM, Boltshauser E, Sayer JA, Gunay-Aygun M, Valente EM, Doherty D. Healthcare recommendations for Joubert syndrome. Am J Med Genet A 2020;182:229–49. 10.1002/ajmg.a.61399 - DOI - PMC - PubMed
-
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, Yachnis AT, Creel G, Frerking B. "Joubert syndrome" revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 1997;12:423–30. 10.1177/088307389701200703 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous