

Dominant clade-featured SARS-CoV-2 co-occurring mutations reveal plausible epistasis: An in silico based hypothetical model
- PMID: 34676891
- PMCID: PMC8661685
- DOI: 10.1002/jmv.27416
Dominant clade-featured SARS-CoV-2 co-occurring mutations reveal plausible epistasis: An in silico based hypothetical model
Erratum in
-
Corrigendum.J Med Virol. 2022 Jul;94(7):3469. doi: 10.1002/jmv.27733. Epub 2022 Apr 11. J Med Virol. 2022. PMID: 35373349 Free PMC article. No abstract available.
Abstract
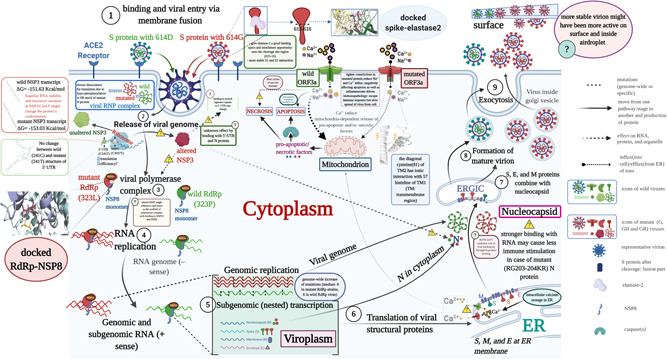
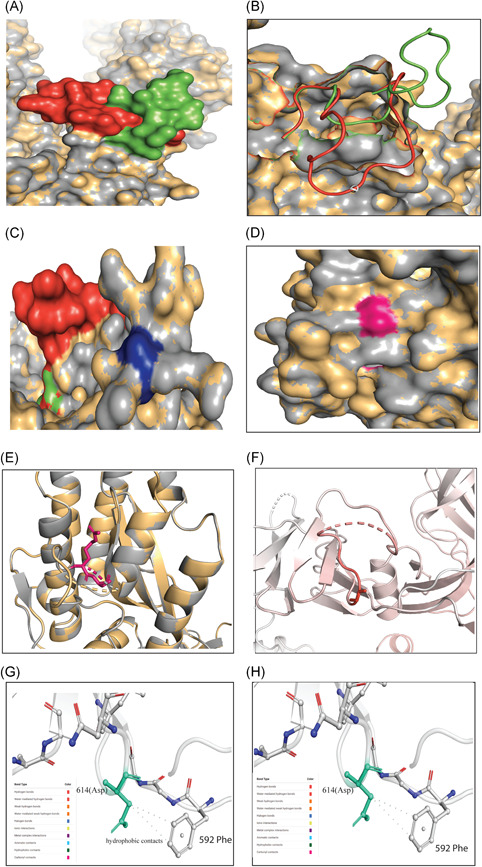
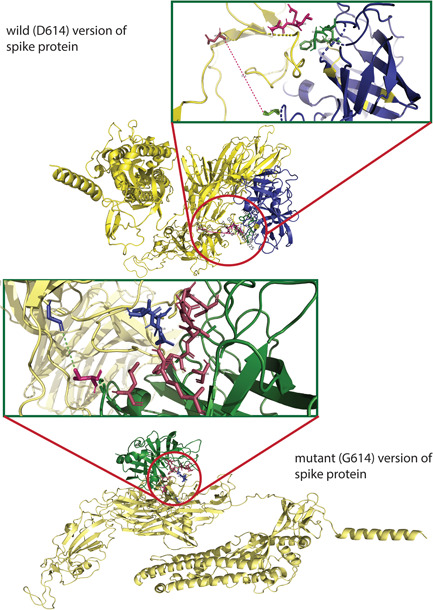
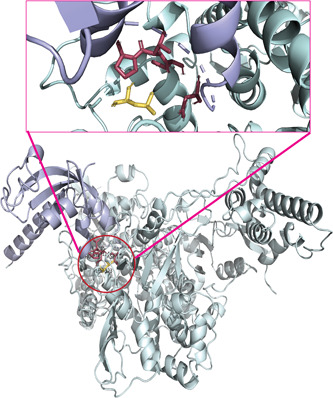
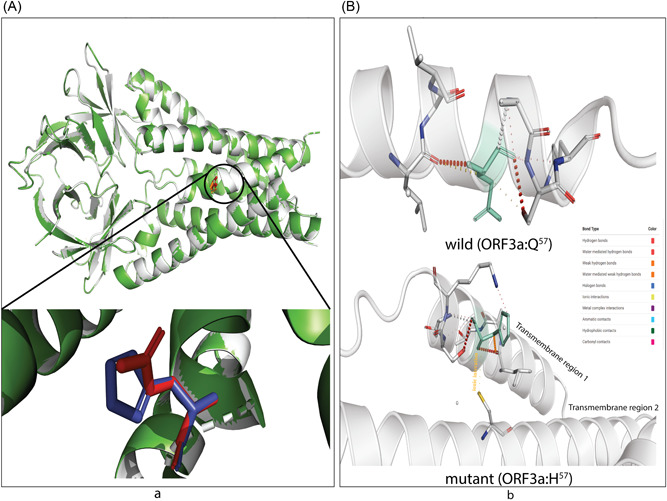
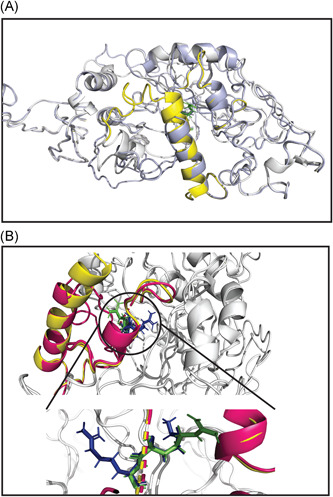
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has evolved into eight fundamental clades with four of these clades (G, GH, GR, and GV) globally prevalent in 2020. To explain plausible epistatic effects of the signature co-occurring mutations of these circulating clades on viral replication and transmission fitness, we proposed a hypothetical model using in silico approach. Molecular docking and dynamics analyses showed the higher infectiousness of a spike mutant through more favorable binding of G614 with the elastase-2. RdRp mutation p.P323L significantly increased genome-wide mutations (p < 0.0001), allowing for more flexible RdRp (mutated)-NSP8 interaction that may accelerate replication. Superior RNA stability and structural variation at NSP3:C241T might impact protein, RNA interactions, or both. Another silent 5'-UTR:C241T mutation might affect translational efficiency and viral packaging. These four G-clade-featured co-occurring mutations might increase viral replication. Sentinel GH-clade ORF3a:p.Q57H variants constricted the ion-channel through intertransmembrane-domain interaction of cysteine(C81)-histidine(H57). The GR-clade N:p.RG203-204KR would stabilize RNA interaction by a more flexible and hypo-phosphorylated SR-rich region. GV-clade viruses seemingly gained the evolutionary advantage of the confounding factors; nevertheless, N:p.A220V might modulate RNA binding with no phenotypic effect. Our hypothetical model needs further retrospective and prospective studies to understand detailed molecular events and their relationship to the fitness of SARS-CoV-2.
Keywords: COVID-19; SARS-CoV-2; clades; co-occurring mutations; fitness; infection paradox; virulence.
© 2021 Wiley Periodicals LLC.
Conflict of interest statement
The authors declare that there are no conflict of interests.
Figures
Similar articles
-
Global variation in SARS-CoV-2 proteome and its implication in pre-lockdown emergence and dissemination of 5 dominant SARS-CoV-2 clades.Infect Genet Evol. 2021 Sep;93:104973. doi: 10.1016/j.meegid.2021.104973. Epub 2021 Jun 18. Infect Genet Evol. 2021. PMID: 34147651 Free PMC article.
-
Structural Mapping of Mutations in Spike, RdRp and Orf3a Genes of SARS-CoV-2 in Influenza Like Illness (ILI) Patients.Viruses. 2021 Jan 19;13(1):136. doi: 10.3390/v13010136. Viruses. 2021. PMID: 33477951 Free PMC article.
-
Time Series Analysis of SARS-CoV-2 Genomes and Correlations among Highly Prevalent Mutations.Microbiol Spectr. 2022 Oct 26;10(5):e0121922. doi: 10.1128/spectrum.01219-22. Epub 2022 Sep 7. Microbiol Spectr. 2022. PMID: 36069583 Free PMC article.
-
Higher entropy observed in SARS-CoV-2 genomes from the first COVID-19 wave in Pakistan.PLoS One. 2021 Aug 31;16(8):e0256451. doi: 10.1371/journal.pone.0256451. eCollection 2021. PLoS One. 2021. PMID: 34464419 Free PMC article.
-
Different selection dynamics of S and RdRp between SARS-CoV-2 genomes with and without the dominant mutations.Infect Genet Evol. 2021 Jul;91:104796. doi: 10.1016/j.meegid.2021.104796. Epub 2021 Mar 3. Infect Genet Evol. 2021. PMID: 33667722 Free PMC article.
Cited by
-
Genomic Analysis of SARS-CoV-2 Alpha, Beta and Delta Variants of Concern Uncovers Signatures of Neutral and Non-Neutral Evolution.Viruses. 2022 Oct 27;14(11):2375. doi: 10.3390/v14112375. Viruses. 2022. PMID: 36366473 Free PMC article.
-
Characterization of SARS-CoV-2 Variants in Military and Civilian Personnel of an Air Force Airport during Three Pandemic Waves in Italy.Microorganisms. 2023 Nov 5;11(11):2711. doi: 10.3390/microorganisms11112711. Microorganisms. 2023. PMID: 38004723 Free PMC article.
-
Modular characterization of SARS-CoV-2 nucleocapsid protein domain functions in nucleocapsid-like assembly.Mol Biomed. 2023 May 22;4(1):16. doi: 10.1186/s43556-023-00129-z. Mol Biomed. 2023. PMID: 37211575 Free PMC article.
-
Corrigendum.J Med Virol. 2022 Jul;94(7):3469. doi: 10.1002/jmv.27733. Epub 2022 Apr 11. J Med Virol. 2022. PMID: 35373349 Free PMC article. No abstract available.
-
Activity and cellular distribution of ORF3a mutants of SARS-CoV-2 variants of concern.J Gen Virol. 2025 Aug;106(8):002135. doi: 10.1099/jgv.0.002135. J Gen Virol. 2025. PMID: 40768270 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
