

Preventing amyotrophic lateral sclerosis: insights from pre-symptomatic neurodegenerative diseases
- PMID: 34677606
- PMCID: PMC8967095
- DOI: 10.1093/brain/awab404
Preventing amyotrophic lateral sclerosis: insights from pre-symptomatic neurodegenerative diseases
Abstract
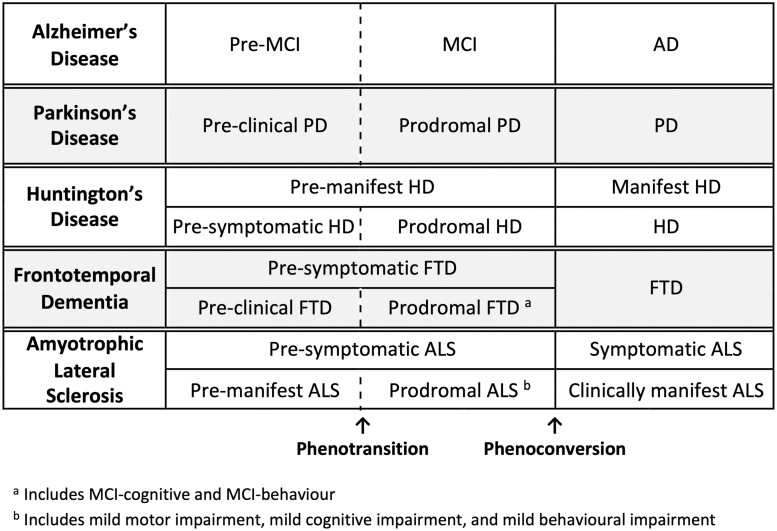
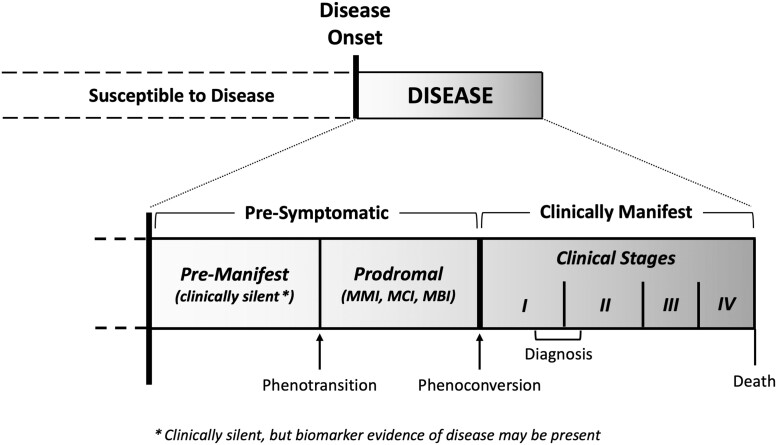
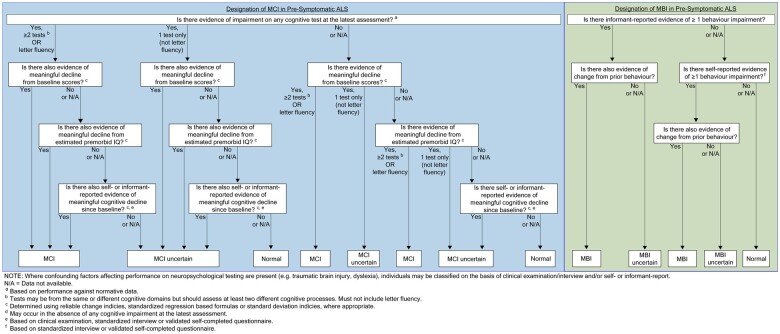
Significant progress has been made in understanding the pre-symptomatic phase of amyotrophic lateral sclerosis. While much is still unknown, advances in other neurodegenerative diseases offer valuable insights. Indeed, it is increasingly clear that the well-recognized clinical syndromes of Alzheimer's disease, Parkinson's disease, Huntington's disease, spinal muscular atrophy and frontotemporal dementia are also each preceded by a pre-symptomatic or prodromal period of varying duration, during which the underlying disease process unfolds, with associated compensatory changes and loss of inherent system redundancy. Key insights from these diseases highlight opportunities for discovery in amyotrophic lateral sclerosis. The development of biomarkers reflecting amyloid and tau has led to a shift in defining Alzheimer's disease based on inferred underlying histopathology. Parkinson's disease is unique among neurodegenerative diseases in the number and diversity of non-genetic biomarkers of pre-symptomatic disease, most notably REM sleep behaviour disorder. Huntington's disease benefits from an ability to predict the likely timing of clinically manifest disease based on age and CAG-repeat length alongside reliable neuroimaging markers of atrophy. Spinal muscular atrophy clinical trials have highlighted the transformational value of early therapeutic intervention, and studies in frontotemporal dementia illustrate the differential role of biomarkers based on genotype. Similar advances in amyotrophic lateral sclerosis would transform our understanding of key events in pathogenesis, thereby dramatically accelerating progress towards disease prevention. Deciphering the biology of pre-symptomatic amyotrophic lateral sclerosis relies on a clear conceptual framework for defining the earliest stages of disease. Clinically manifest amyotrophic lateral sclerosis may emerge abruptly, especially among those who harbour genetic mutations associated with rapidly progressive amyotrophic lateral sclerosis. However, the disease may also evolve more gradually, revealing a prodromal period of mild motor impairment preceding phenoconversion to clinically manifest disease. Similarly, cognitive and behavioural impairment, when present, may emerge gradually, evolving through a prodromal period of mild cognitive impairment or mild behavioural impairment before progression to amyotrophic lateral sclerosis. Biomarkers are critically important to studying pre-symptomatic amyotrophic lateral sclerosis and essential to efforts to intervene therapeutically before clinically manifest disease emerges. The use of non-genetic biomarkers, however, presents challenges related to counselling, informed consent, communication of results and limited protections afforded by existing legislation. Experiences from pre-symptomatic genetic testing and counselling, and the legal protections against discrimination based on genetic data, may serve as a guide. Building on what we have learned-more broadly from other pre-symptomatic neurodegenerative diseases and specifically from amyotrophic lateral sclerosis gene mutation carriers-we present a road map to early intervention, and perhaps even disease prevention, for all forms of amyotrophic lateral sclerosis.
Keywords: amyotrophic lateral sclerosis (ALS); disease prevention; neurodegeneration; pre-symptomatic.
© The Author(s) (2021). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Comment in
-
Pre-symptomatic spinal muscular atrophy: a proposed nosology.Brain. 2022 Jul 29;145(7):2247-2249. doi: 10.1093/brain/awac125. Brain. 2022. PMID: 35429273 No abstract available.
References
-
- Rabin LA, Smart CM, Amariglio RE.. Subjective cognitive decline in preclinical Alzheimer's disease. Annu Rev Clin Psychol. 2017;13:369–396. - PubMed
Publication types
MeSH terms
Grants and funding
- ALCHALABI-DOBSON/APR14/829-791/MNDA_/Motor Neurone Disease Association/United Kingdom
- K24 AG045333/AG/NIA NIH HHS/United States
- G0600974/MRC_/Medical Research Council/United Kingdom
- TURNER/JAN13/944-795/MNDA_/Motor Neurone Disease Association/United Kingdom
- ALCHALABI-TALBOT/APR14/926-794/MNDA_/Motor Neurone Disease Association/United Kingdom
- MR/J009482/1/MRC_/Medical Research Council/United Kingdom
- MR/M023664/1/MRC_/Medical Research Council/United Kingdom
- P30 AG062677/AG/NIA NIH HHS/United States
- R01 NS105479/NS/NINDS NIH HHS/United States
- MR/R024804/1/MRC_/Medical Research Council/United Kingdom
- MR/T046015/1/MRC_/Medical Research Council/United Kingdom
- R01 NS086452/NS/NINDS NIH HHS/United States
- MR/L501529/1/MRC_/Medical Research Council/United Kingdom
- K01 AG057796/AG/NIA NIH HHS/United States
- U54 NS092091/NS/NINDS NIH HHS/United States
- P01 AG019724/AG/NIA NIH HHS/United States
- TURNER/OCT18/989-797/MNDA_/Motor Neurone Disease Association/United Kingdom
- MR/M008525/1/MRC_/Medical Research Council/United Kingdom
- U01 AG006786/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
