

Functions predict horizontal gene transfer and the emergence of antibiotic resistance
- PMID: 34678056
- PMCID: PMC8535800
- DOI: 10.1126/sciadv.abj5056
Functions predict horizontal gene transfer and the emergence of antibiotic resistance
Abstract
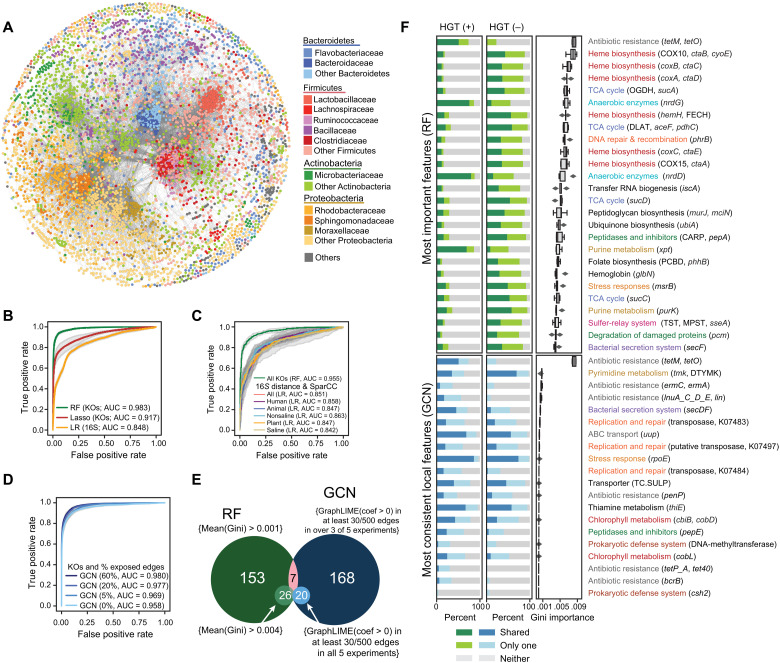
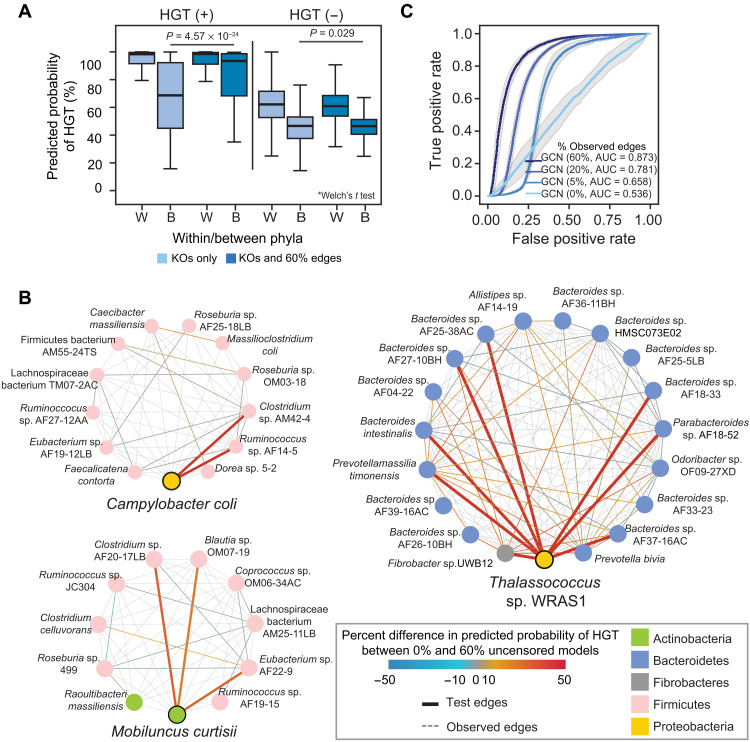
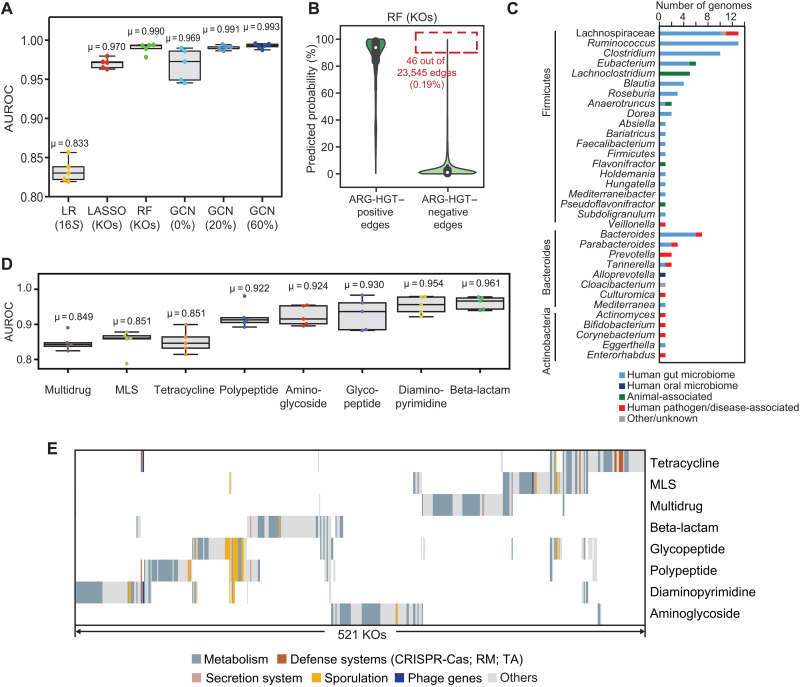
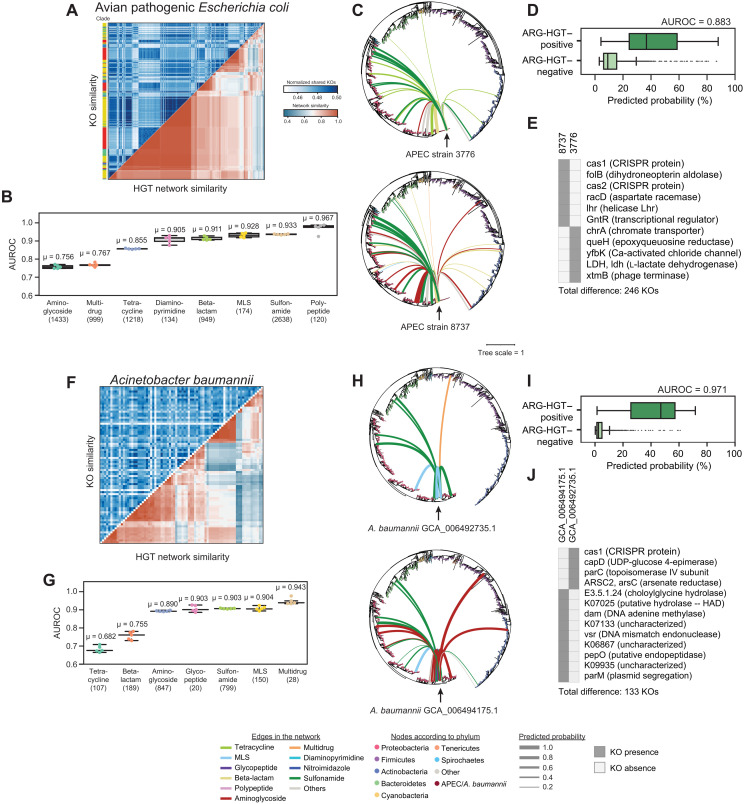
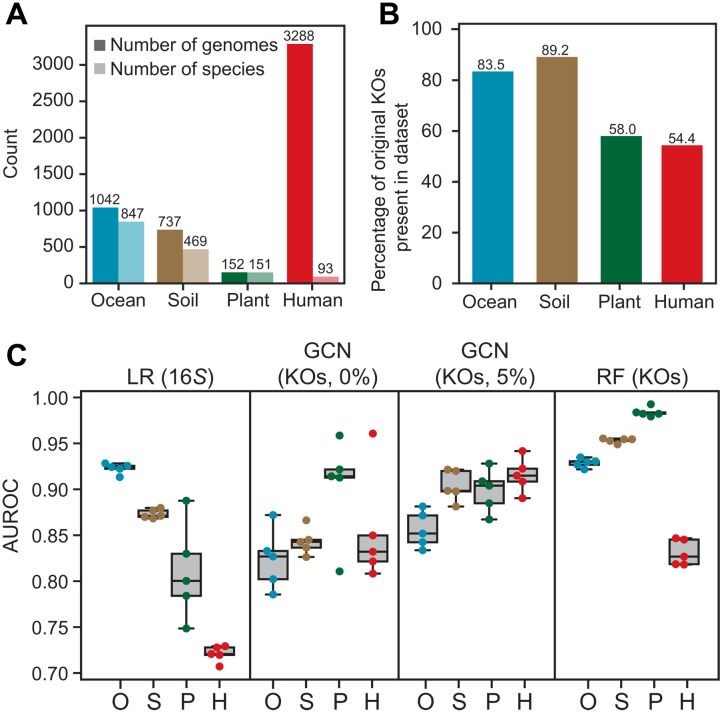
Phylogenetic distance, shared ecology, and genomic constraints are often cited as key drivers governing horizontal gene transfer (HGT), although their relative contributions are unclear. Here, we apply machine learning algorithms to a curated set of diverse bacterial genomes to tease apart the importance of specific functional traits on recent HGT events. We find that functional content accurately predicts the HGT network [area under the receiver operating characteristic curve (AUROC) = 0.983], and performance improves further (AUROC = 0.990) for transfers involving antibiotic resistance genes (ARGs), highlighting the importance of HGT machinery, niche-specific, and metabolic functions. We find that high-probability not-yet detected ARG transfer events are almost exclusive to human-associated bacteria. Our approach is robust at predicting the HGT networks of pathogens, including Acinetobacter baumannii and Escherichia coli, as well as within localized environments, such as an individual’s gut microbiome.
Figures
References
-
- Smillie C. S., Smith M. B., Friedman J., Cordero O. X., David L. A., Alm E. J., Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480, 241–244 (2011). - PubMed
-
- Beaulaurier J., Zhu S., Deikus G., Mogno I., Zhang X.-S., Davis-Richardson A., Canepa R., Triplett E. W., Faith J. J., Sebra R., Schadt E. E., Fang G., Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat. Biotechnol. 36, 61–69 (2018). - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
