

Osteogenesis Imperfecta: Current and Prospective Therapies
- PMID: 34680126
- PMCID: PMC8533546
- DOI: 10.3390/biom11101493
Osteogenesis Imperfecta: Current and Prospective Therapies
Abstract
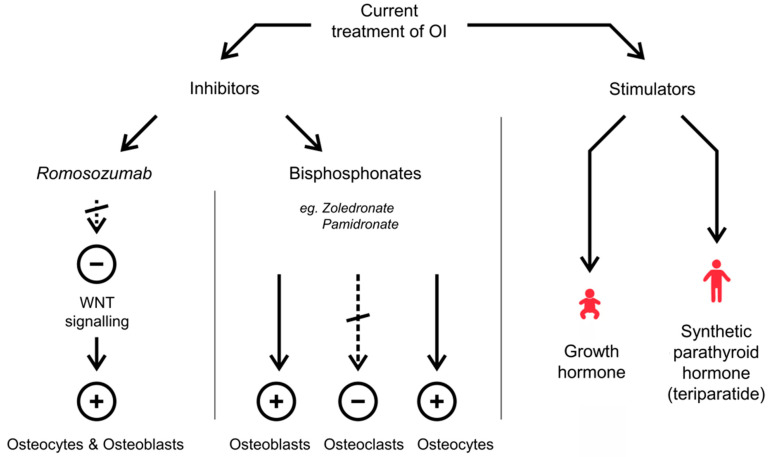
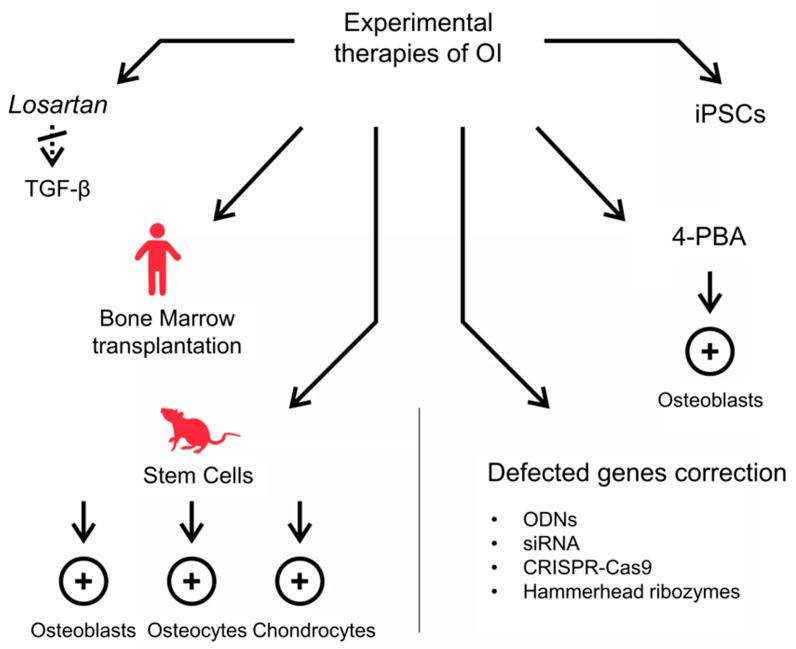
Osteogenesis Imperfecta (OI) is a group of connective tissue disorders with a broad range of phenotypes characterized primarily by bone fragility. The prevalence of OI ranges from about 1:15,000 to 1:20,000 births. Five types of the disease are commonly distinguished, ranging from a mild (type I) to a lethal one (type II). Types III and IV are severe forms allowing survival after the neonatal period, while type V is characterized by a mild to moderate phenotype with calcification of interosseous membranes. In most cases, there is a reduction in the production of normal type I collagen (col I) or the synthesis of abnormal collagen as a result of mutations in col I genes. Moreover, mutations in genes involved in col I synthesis and processing as well as in osteoblast differentiation have been reported. The currently available treatments try to prevent fractures, control symptoms and increase bone mass. Commonly used medications in OI treatment are bisphosphonates, Denosumab, synthetic parathyroid hormone and growth hormone for children therapy. The main disadvantages of these therapies are their relatively weak effectiveness, lack of effects in some patients or cytotoxic side effects. Experimental approaches, particularly those based on stem cell transplantation and genetic engineering, seem to be promising to improve the therapeutic effects of OI.
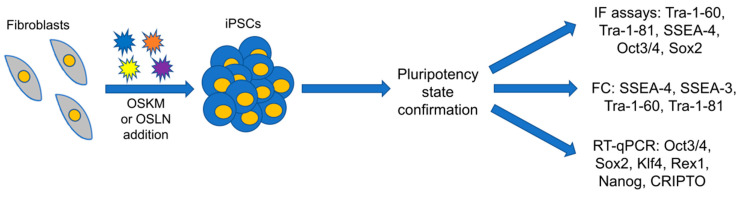
Keywords: gene therapy; iPSCs; mesenchymal stem cells; osteogenesis imperfecta; treatment.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Mortier G.R., Cohn D.H., Cormier-Daire V., Hall C., Krakow D., Mundlos S., Nishimura G., Robertson S., Sangiorgi L., Savarirayan R., et al. Nosology and classification of genetic skeletal disorders: 2019 revision. [(accessed on 2 November 2019)];Am. J. Med. Genet. Part A. 2019 179:2393–2419. doi: 10.1002/ajmg.a.61366. Available online: http://www.ncbi.nlm.nih.gov/pubmed/31633310. - DOI - PubMed
-
- Landis W.J., Hodgens K.J., Song M.J., Arena J., Kiyonaga S., Marko M., Owen C., McEwen B.F. Mineralization of collagen may occur on fibril surfaces: Evidence from conven-tional and high-voltage electron microscopy and three-dimensional imaging. J. Struct. Biol. 1996;117:24–35. doi: 10.1006/jsbi.1996.0066. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
