

Exploring Semi-Quantitative Metagenomic Studies Using Oxford Nanopore Sequencing: A Computational and Experimental Protocol
- PMID: 34680891
- PMCID: PMC8536095
- DOI: 10.3390/genes12101496
Exploring Semi-Quantitative Metagenomic Studies Using Oxford Nanopore Sequencing: A Computational and Experimental Protocol
Abstract
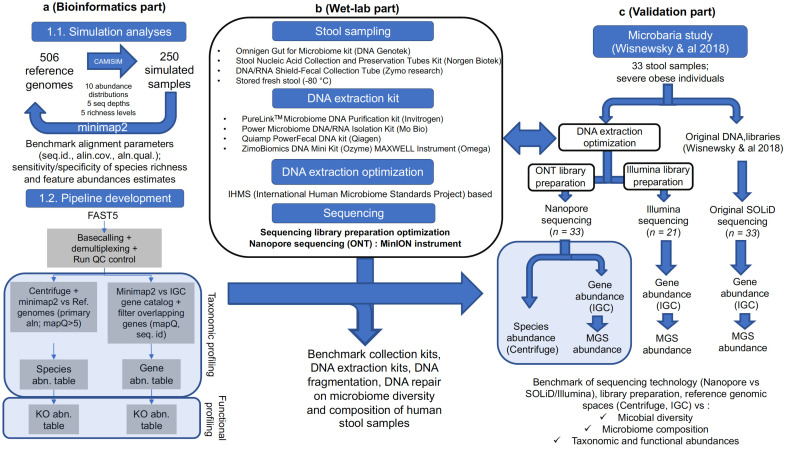
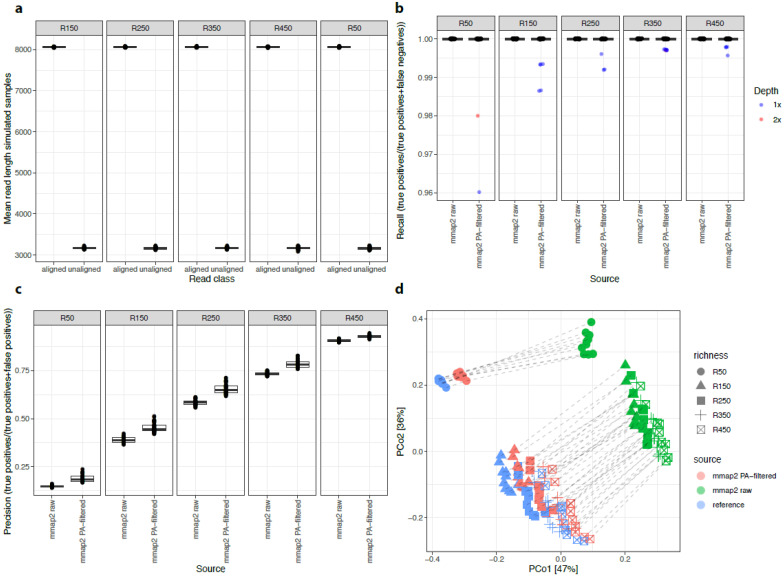
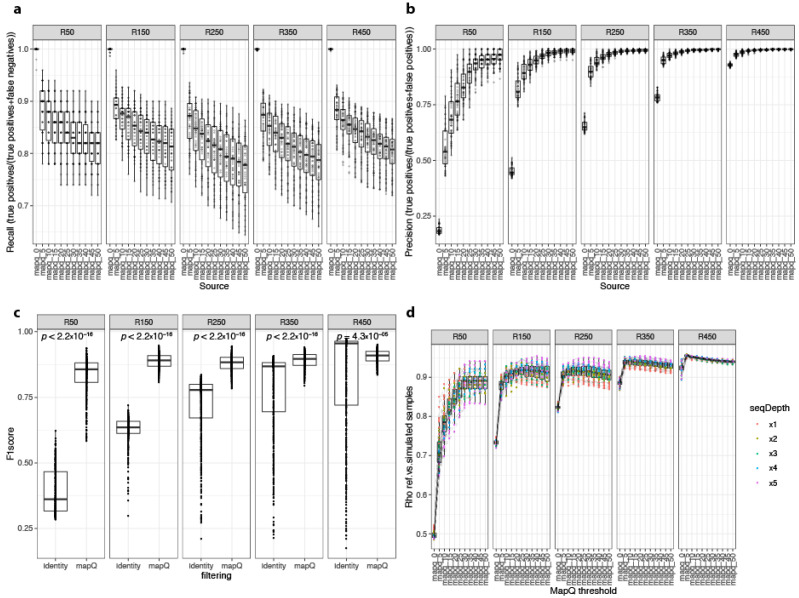
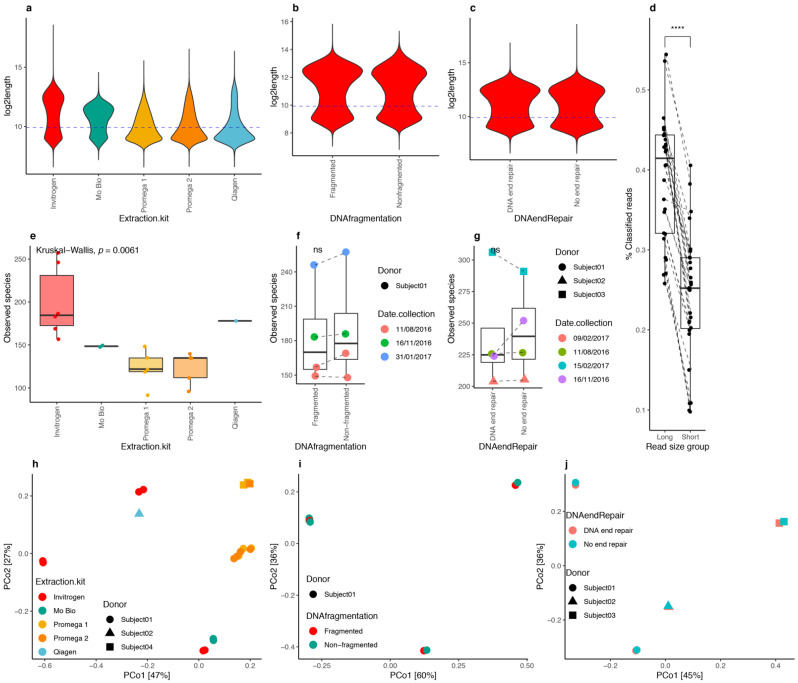
The gut microbiome plays a major role in chronic diseases, of which several are characterized by an altered composition and diversity of bacterial communities. Large-scale sequencing projects allowed for characterizing the perturbations of these communities. However, translating these discoveries into clinical applications remains a challenge. To facilitate routine implementation of microbiome profiling in clinical settings, portable, real-time, and low-cost sequencing technologies are needed. Here, we propose a computational and experimental protocol for whole-genome semi-quantitative metagenomic studies of human gut microbiome with Oxford Nanopore sequencing technology (ONT) that could be applied to other microbial ecosystems. We developed a bioinformatics protocol to analyze ONT sequences taxonomically and functionally and optimized preanalytic protocols, including stool collection and DNA extraction methods to maximize read length. This is a critical parameter for the sequence alignment and classification. Our protocol was evaluated using simulations of metagenomic communities, which reflect naturally occurring compositional variations. Next, we validated both protocols using stool samples from a bariatric surgery cohort, sequenced with ONT, Illumina, and SOLiD technologies. Results revealed similar diversity and microbial composition profiles. This protocol can be implemented in a clinical or research setting, bringing rapid personalized whole-genome profiling of target microbiome species.
Keywords: MinION; Oxford Nanopore Technologies; gut microbiota; microbial DNA extraction; microbiome; obesity; semi-quantitative metagenomics; sequencing; simulation.
Conflict of interest statement
K.C. is a consultant for Danone Research, LNC Therapeutics, and Confo Therapeutics for work unassociated with the present study. J.-D.Z. is a consultant for Quinten for work unassociated with the present study.
Figures
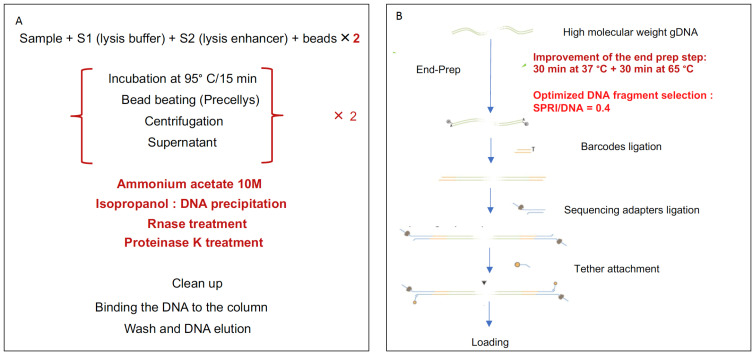
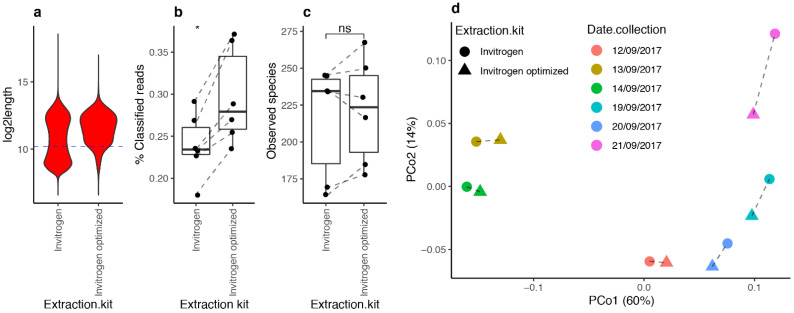
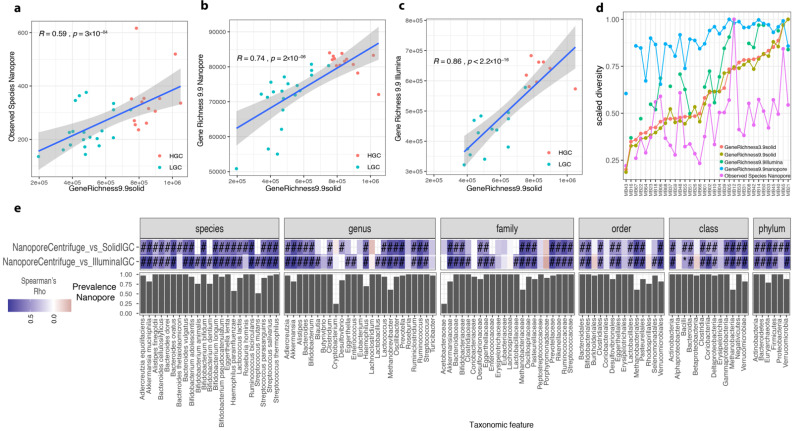
References
-
- Vieira-Silva S., Falony G., Belda E., Nielsen T., Aron-Wisnewsky J., Chakaroun R., Forslund S.K., Assmann K., Valles-Colomer M., Nguyen T.T.D., et al. Statin Therapy Is Associated with Lower Prevalence of Gut Microbiota Dysbiosis. Nature. 2020;581:310–315. doi: 10.1038/s41586-020-2269-x. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
