

Improved SNV Discovery in Barcode-Stratified scRNA-seq Alignments
- PMID: 34680953
- PMCID: PMC8535975
- DOI: 10.3390/genes12101558
Improved SNV Discovery in Barcode-Stratified scRNA-seq Alignments
Abstract
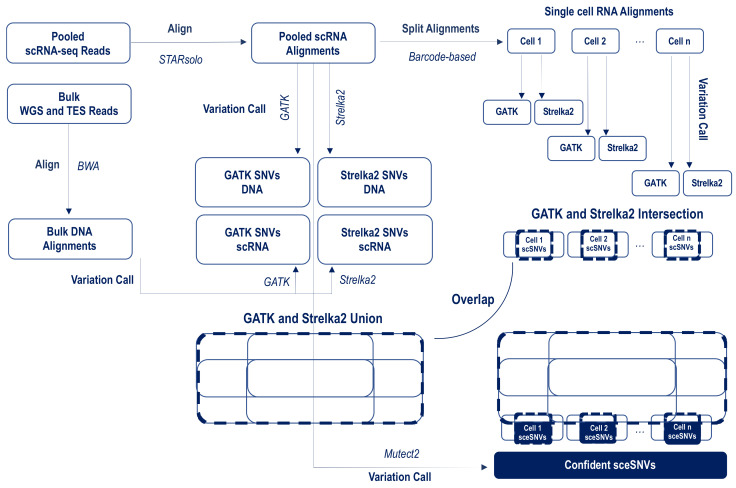
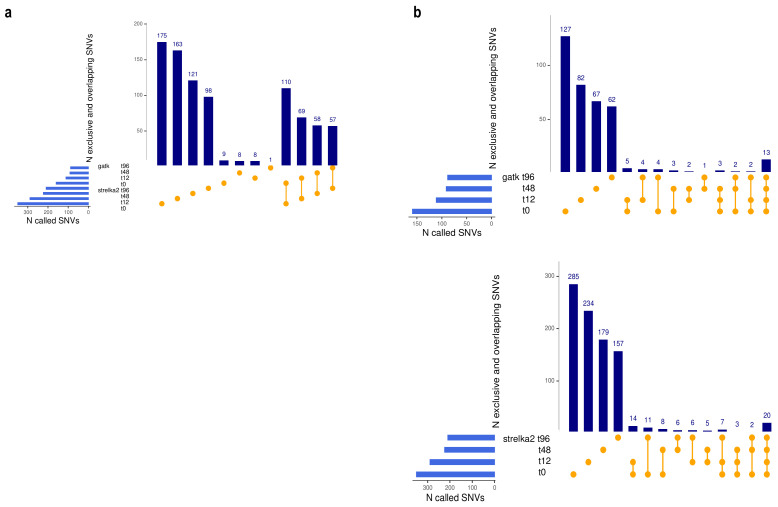
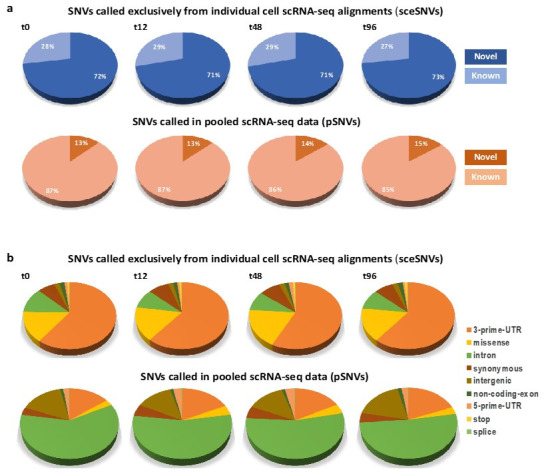
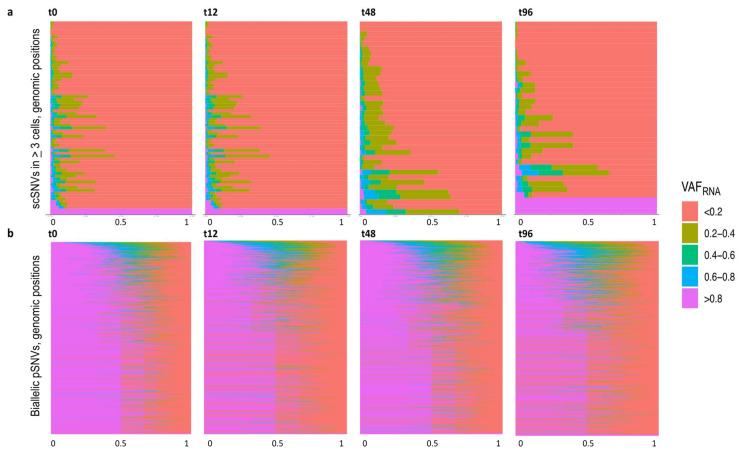
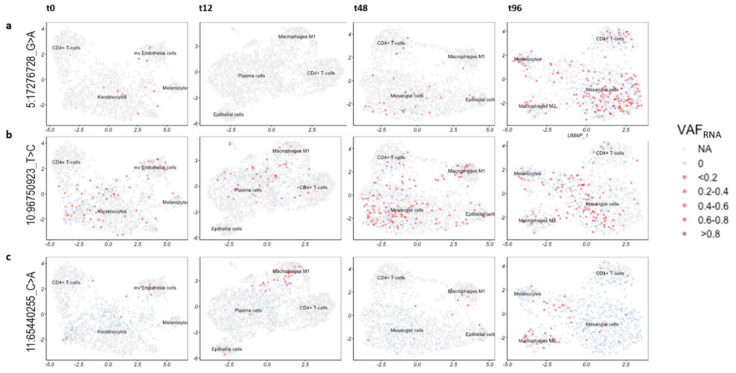
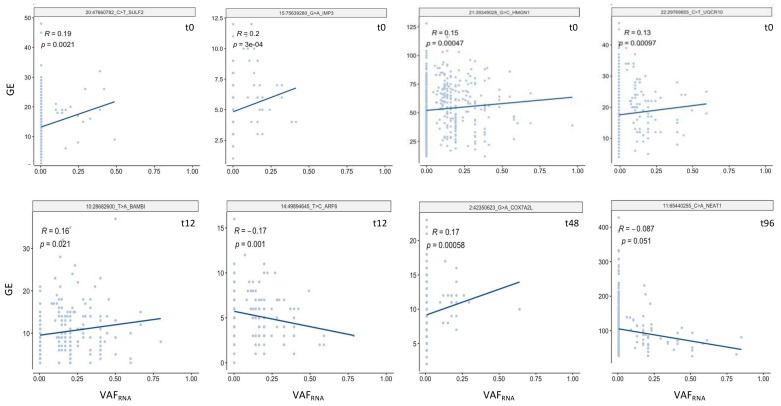
Currently, the detection of single nucleotide variants (SNVs) from 10 x Genomics single-cell RNA sequencing data (scRNA-seq) is typically performed on the pooled sequencing reads across all cells in a sample. Here, we assess the gaining of information regarding SNV assessments from individual cell scRNA-seq data, wherein the alignments are split by cellular barcode prior to the variant call. We also reanalyze publicly available data on the MCF7 cell line during anticancer treatment. We assessed SNV calls by three variant callers-GATK, Strelka2, and Mutect2, in combination with a method for the cell-level tabulation of the sequencing read counts bearing variant alleles-SCReadCounts (single-cell read counts). Our analysis shows that variant calls on individual cell alignments identify at least a two-fold higher number of SNVs as compared to the pooled scRNA-seq; these SNVs are enriched in novel variants and in stop-codon and missense substitutions. Our study indicates an immense potential of SNV calls from individual cell scRNA-seq data and emphasizes the need for cell-level variant detection approaches and tools, which can contribute to the understanding of the cellular heterogeneity and the relationships to phenotypes, and help elucidate somatic mutation evolution and functionality.
Keywords: SNP; SNV; SNV expression; expressed SNVs; mutation; scRNA-seq; somatic mutation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
