

ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer's Disease
- PMID: 34681851
- PMCID: PMC8539681
- DOI: 10.3390/ijms222011185
ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer's Disease
Abstract
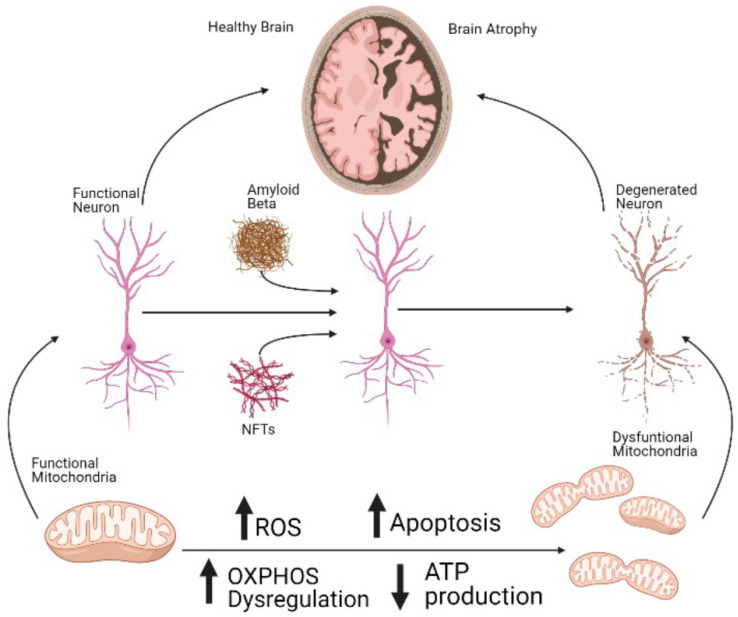
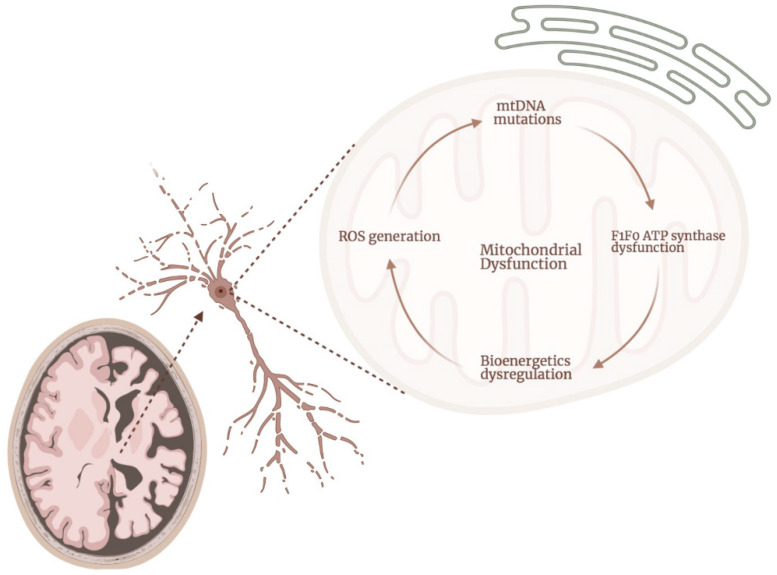
Alzheimer's Disease (AD) is the most common neurodegenerative disorder in our society, as the population ages, its incidence is expected to increase in the coming decades. The etiopathology of this disease still remains largely unclear, probably because of the highly complex and multifactorial nature of AD. However, the presence of mitochondrial dysfunction has been broadly described in AD neurons and other cellular populations within the brain, in a wide variety of models and organisms, including post-mortem humans. Mitochondria are complex organelles that play a crucial role in a wide range of cellular processes, including bioenergetics. In fact, in mammals, including humans, the main source of cellular ATP is the oxidative phosphorylation (OXPHOS), a process that occurs in the mitochondrial electron transfer chain (ETC). The last enzyme of the ETC, and therefore the ulterior generator of ATP, is the ATP synthase. Interestingly, in mammalian cells, the ATP synthase can also degrade ATP under certain conditions (ATPase), which further illustrates the crucial role of this enzyme in the regulation of cellular bioenergetics and metabolism. In this collaborative review, we aim to summarize the knowledge of the presence of dysregulated ATP synthase, and of other components of mammalian mitochondrial bioenergetics, as an early event in AD. This dysregulation can act as a trigger of the dysfunction of the organelle, which is a clear component in the etiopathology of AD. Consequently, the pharmacological modulation of the ATP synthase could be a potential strategy to prevent mitochondrial dysfunction in AD.
Keywords: ATP synthase; ATPase; Alzheimer’s disease; OXPHOS; mitochondria; mitochondrial dysfunction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ten Kate M., Dicks E., Visser P.J., van der Flier W.M., Teunissen C.E., Barkhof F., Scheltens P., Tijms B.M., Alzheimer’s Disease Neuroimaging Initiative Atrophy subtypes in prodromal Alzheimer’s disease are associated with cognitive decline. Brain. 2018;141:3443–3456. doi: 10.1093/brain/awy264. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
