

The flashfm approach for fine-mapping multiple quantitative traits
- PMID: 34686674
- PMCID: PMC8536717
- DOI: 10.1038/s41467-021-26364-y
The flashfm approach for fine-mapping multiple quantitative traits
Abstract
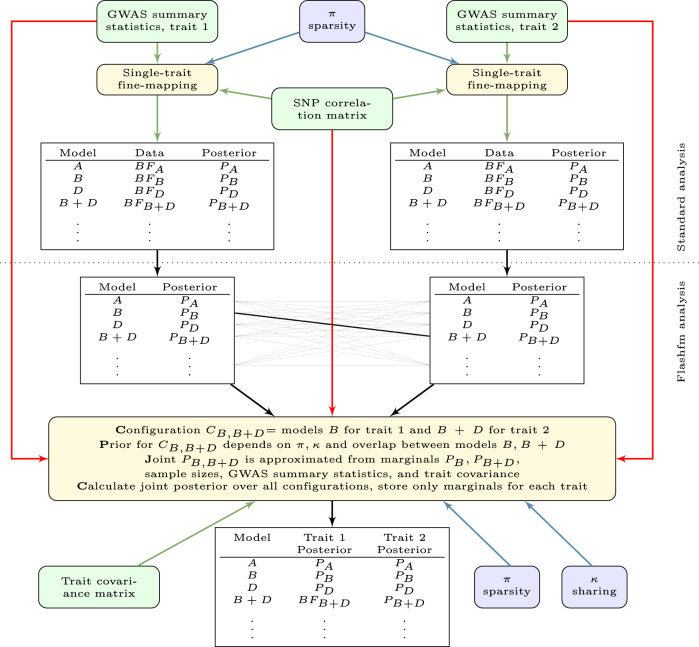
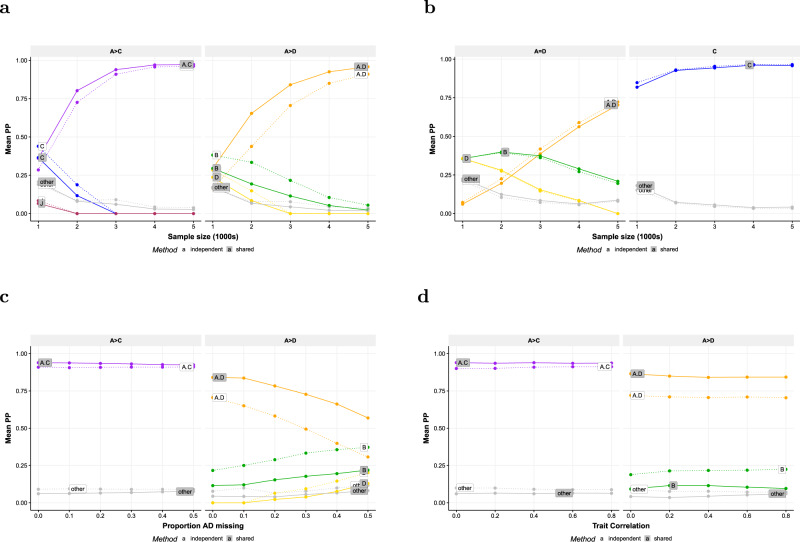
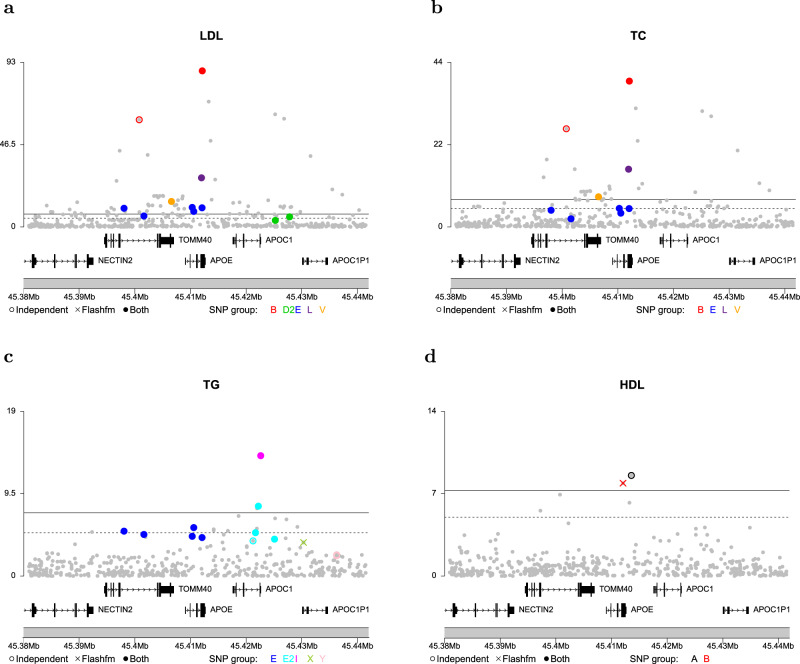
Joint fine-mapping that leverages information between quantitative traits could improve accuracy and resolution over single-trait fine-mapping. Using summary statistics, flashfm (flexible and shared information fine-mapping) fine-maps signals for multiple traits, allowing for missing trait measurements and use of related individuals. In a Bayesian framework, prior model probabilities are formulated to favour model combinations that share causal variants to capitalise on information between traits. Simulation studies demonstrate that both approaches produce broadly equivalent results when traits have no shared causal variants. When traits share at least one causal variant, flashfm reduces the number of potential causal variants by 30% compared with single-trait fine-mapping. In a Ugandan cohort with 33 cardiometabolic traits, flashfm gave a 20% reduction in the total number of potential causal variants from single-trait fine-mapping. Here we show flashfm is computationally efficient and can easily be deployed across publicly available summary statistics for signals in up to six traits.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
