

AdRoit is an accurate and robust method to infer complex transcriptome composition
- PMID: 34686758
- PMCID: PMC8536787
- DOI: 10.1038/s42003-021-02739-1
AdRoit is an accurate and robust method to infer complex transcriptome composition
Abstract
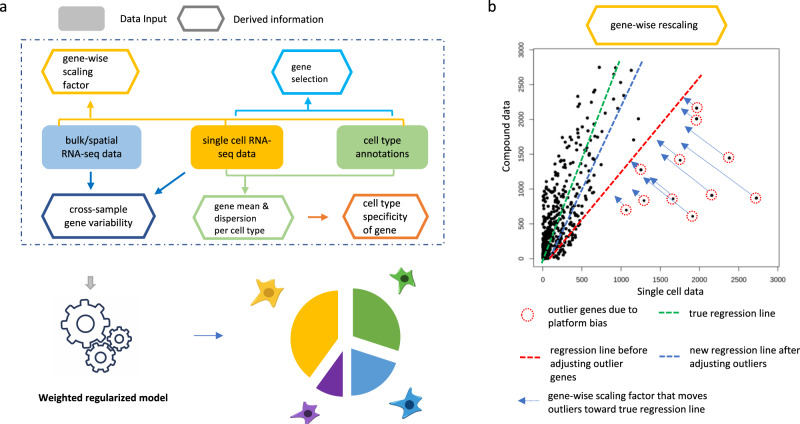
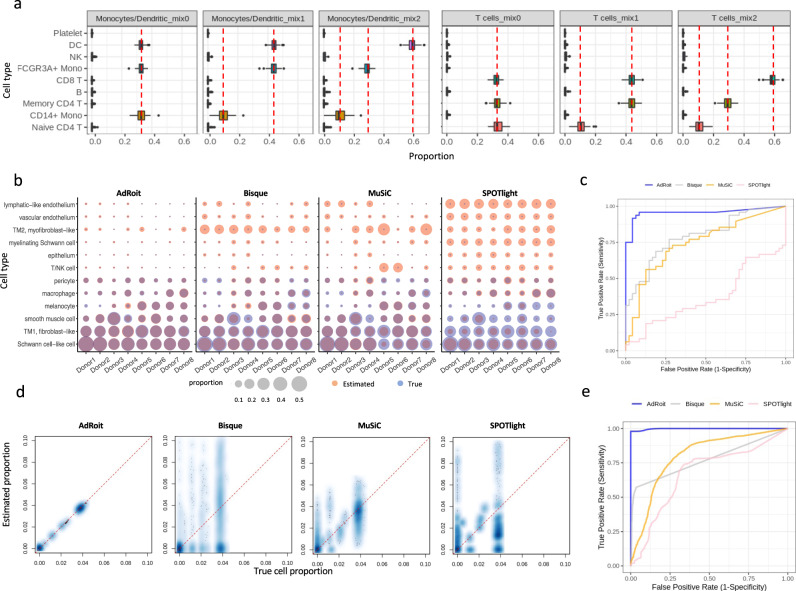
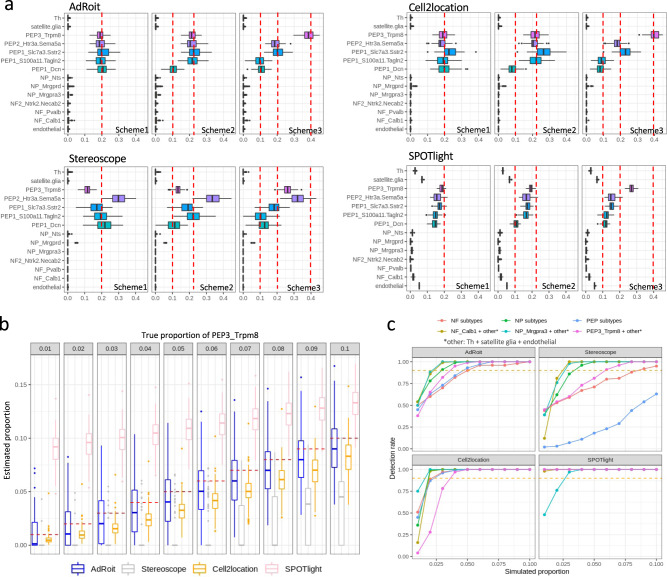
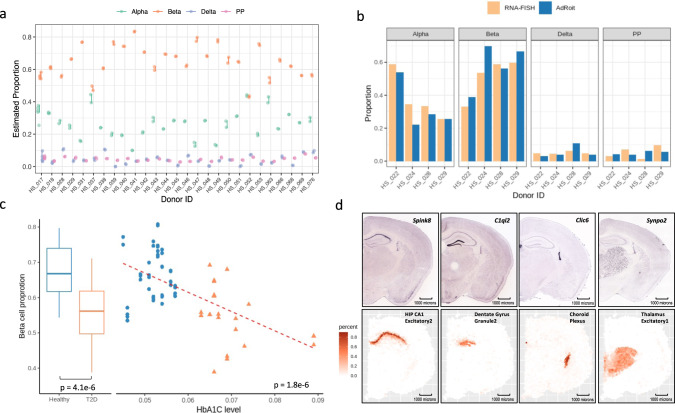
Bulk RNA sequencing provides the opportunity to understand biology at the whole transcriptome level without the prohibitive cost of single cell profiling. Advances in spatial transcriptomics enable to dissect tissue organization and function by genome-wide gene expressions. However, the readout of both technologies is the overall gene expression across potentially many cell types without directly providing the information of cell type constitution. Although several in-silico approaches have been proposed to deconvolute RNA-Seq data composed of multiple cell types, many suffer a deterioration of performance in complex tissues. Here we present AdRoit, an accurate and robust method to infer the cell composition from transcriptome data of mixed cell types. AdRoit uses gene expression profiles obtained from single cell RNA sequencing as a reference. It employs an adaptive learning approach to alleviate the sequencing technique difference between the single cell and the bulk (or spatial) transcriptome data, enhancing cross-platform readout comparability. Our systematic benchmarking and applications, which include deconvoluting complex mixtures that encompass 30 cell types, demonstrate its preferable sensitivity and specificity compared to many existing methods as well as its utilities. In addition, AdRoit is computationally efficient and runs orders of magnitude faster than most methods.
© 2021. The Author(s).
Conflict of interest statement
T.Y., Y.B., W.F., and G.S.A. have filed a patent application relating to the AdRoit computational framework. M.L.-F. is an employee of Cellular Longevity. The remaining authors are employees and shareholders of Regeneron Pharmaceuticals, although the manuscript’s subject matter does not have any relationship to any products or services of this corporation.
Figures


References
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
