

Standardized Workflow for Precise Mid- and High-Throughput Proteomics of Blood Biofluids
- PMID: 34687543
- PMCID: PMC11175165
- DOI: 10.1093/clinchem/hvab202
Standardized Workflow for Precise Mid- and High-Throughput Proteomics of Blood Biofluids
Abstract
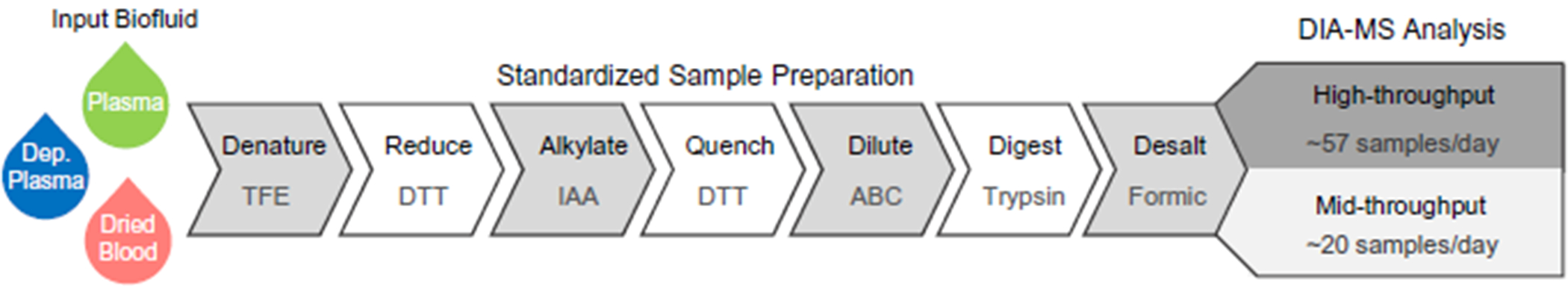
Background: Accurate discovery assay workflows are critical for identifying authentic circulating protein biomarkers in diverse blood matrices. Maximizing the commonalities in the proteomic workflows between different biofluids simplifies the approach and increases the likelihood for reproducibility. We developed a workflow that can accommodate 3 blood-based proteomes: naive plasma, depleted plasma and dried blood.
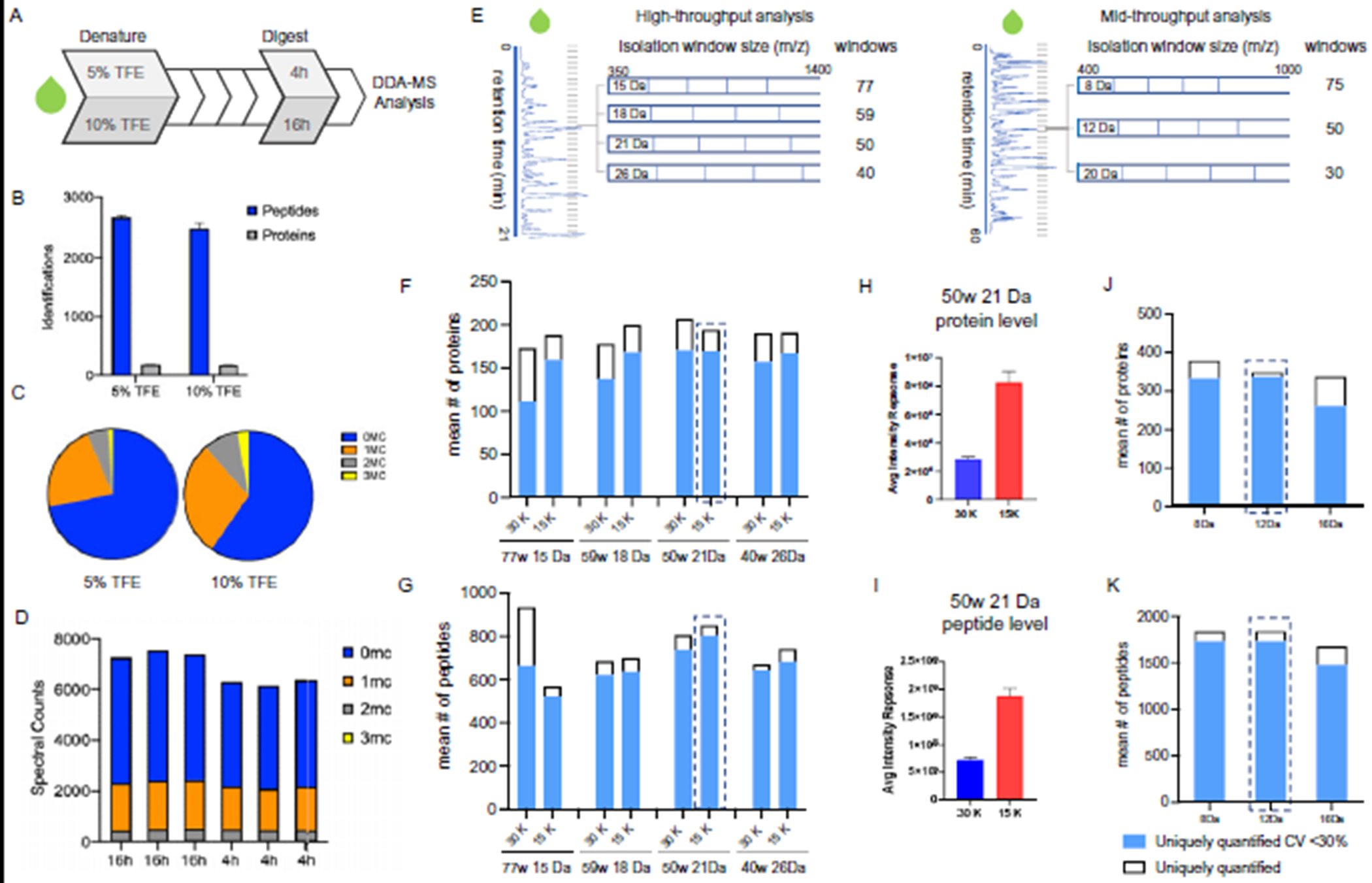
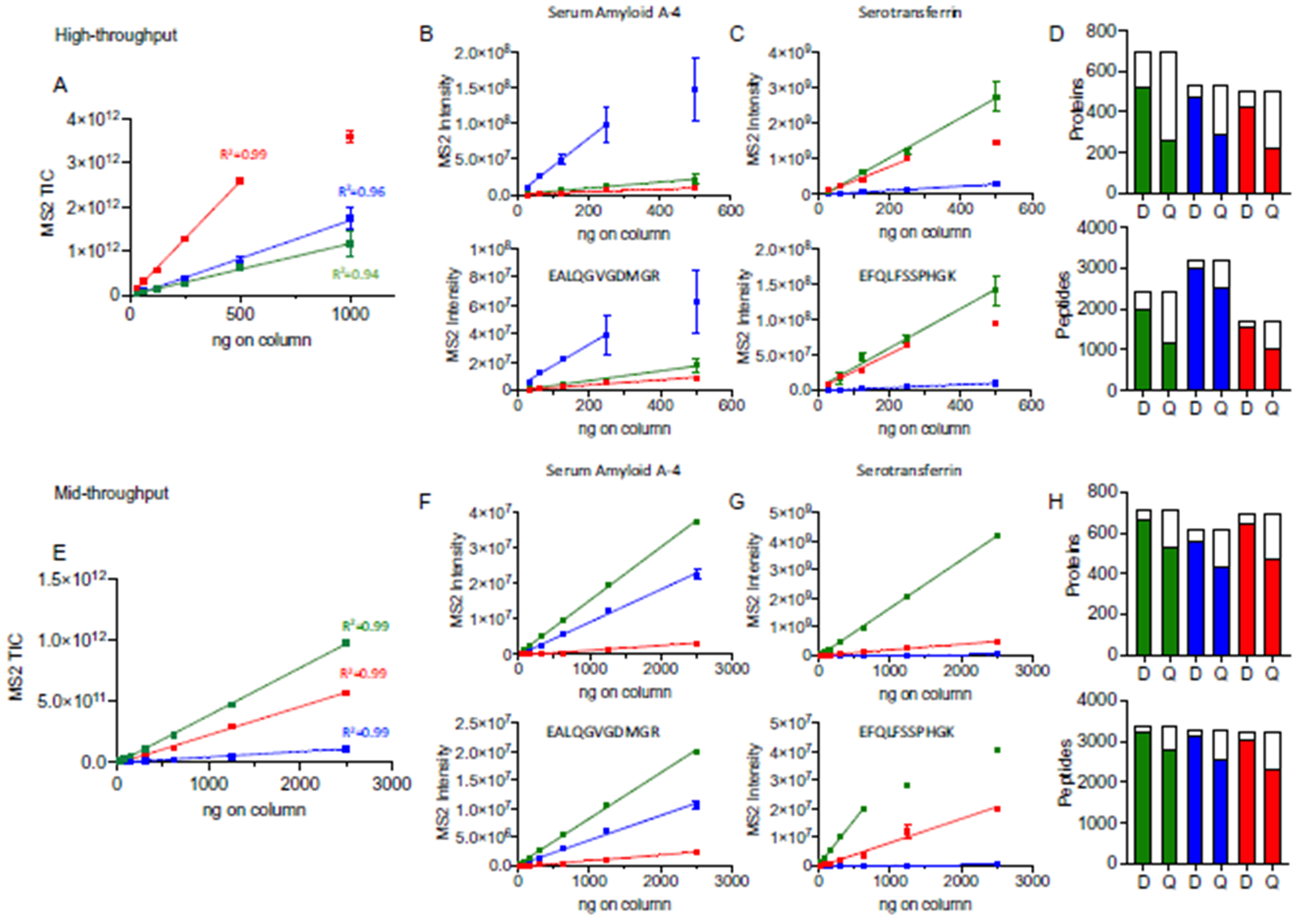
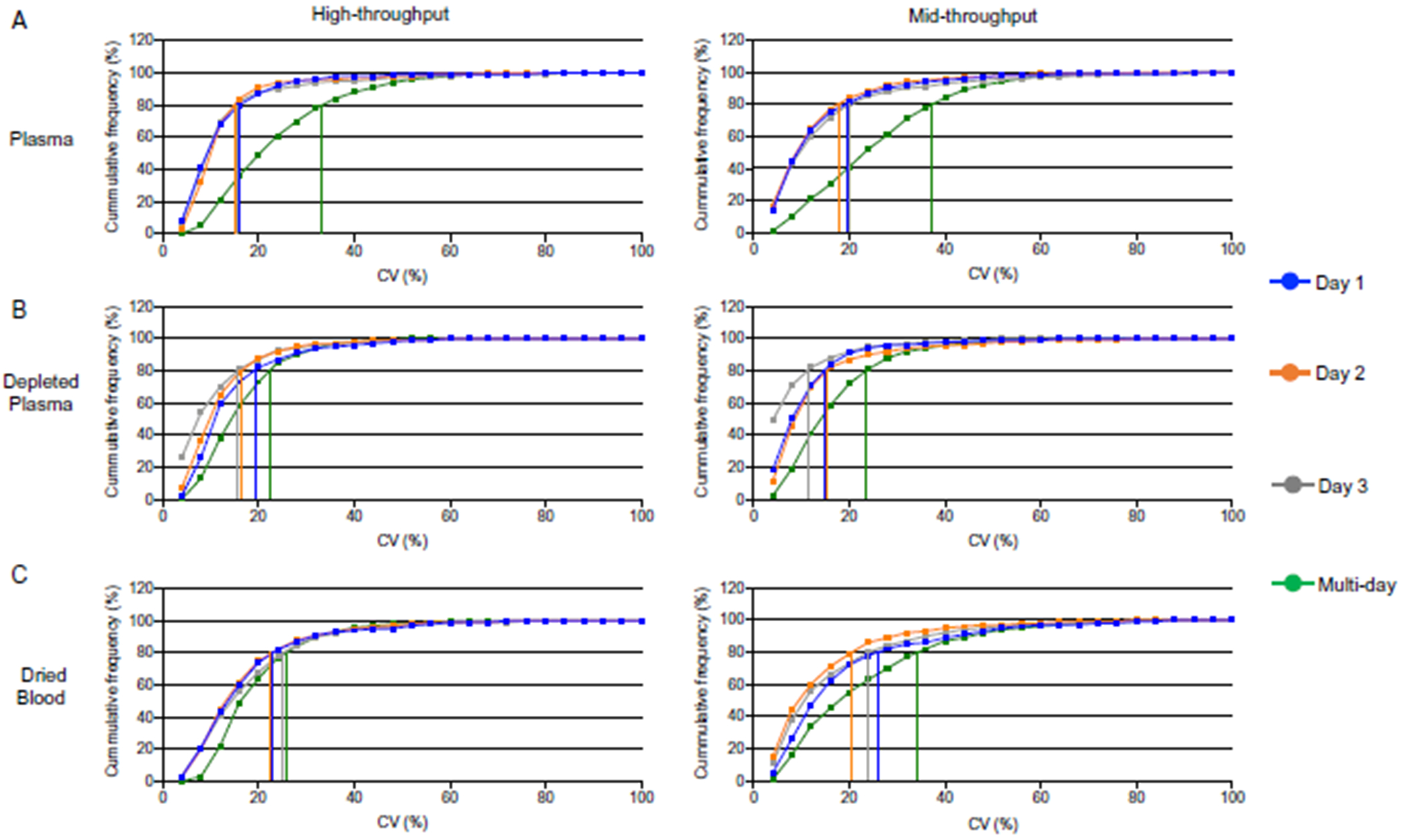
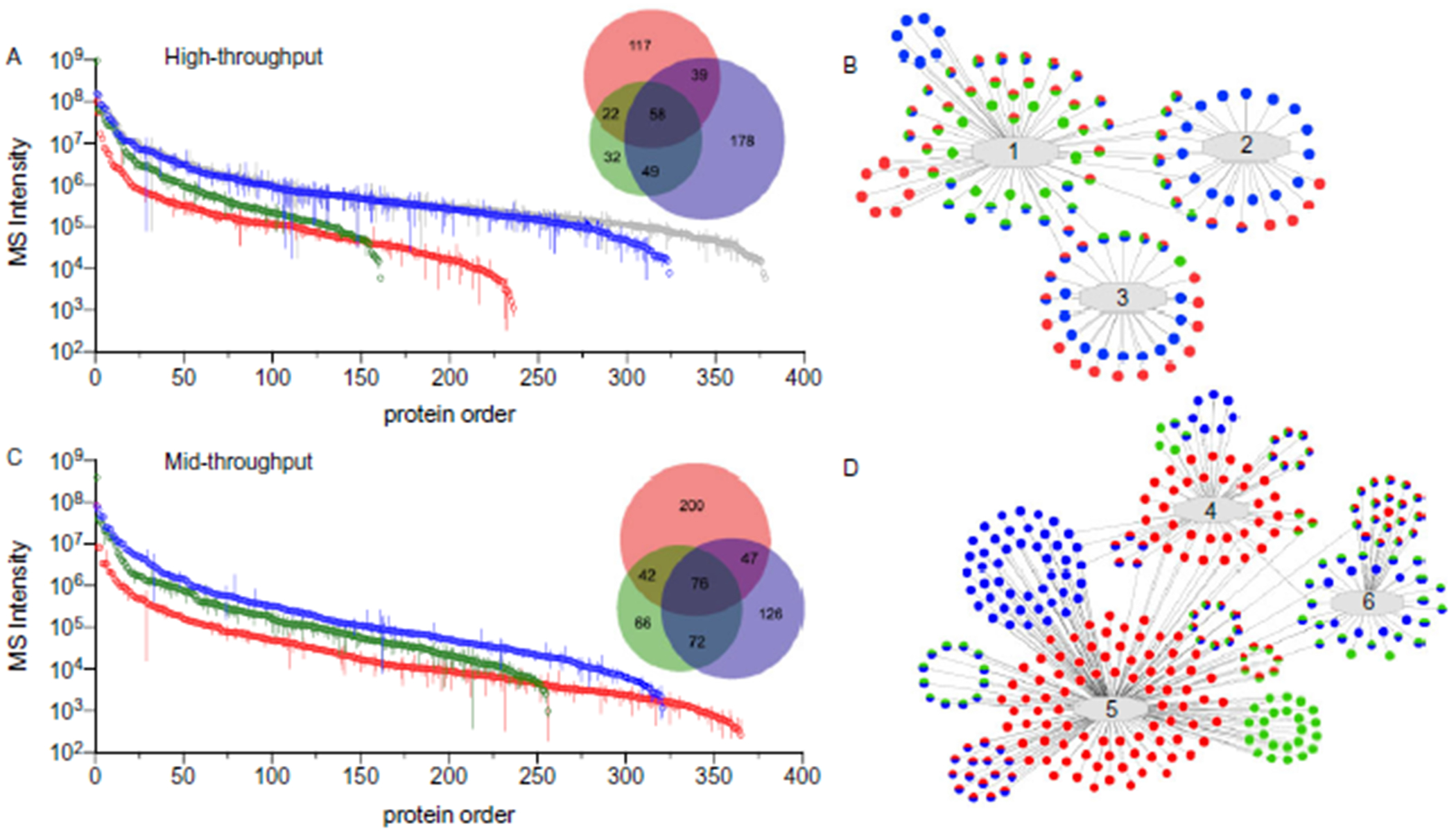
Methods: Optimal conditions for sample preparation and data independent acquisition-mass spectrometry analysis were established in plasma then automated for depleted plasma and dried blood. The mass spectrometry workflow was modified to facilitate sensitive high-throughput analysis or deeper profiling with mid-throughput analysis. Analytical performance was evaluated by the linear response of peptides and proteins to a 6- or 7-point dilution curve and the reproducibility of the relative peptide and protein intensity for 5 digestion replicates per day on 3 different days for each biofluid.
Results: Using the high-throughput workflow, 74% (plasma), 93% (depleted), and 87% (dried blood) displayed an inter-day CV <30%. The mid-throughput workflow had 67% (plasma), 90% (depleted), and 78% (dried blood) of peptides display an inter-day CV <30%. Lower limits of detection and quantification were determined for peptides and proteins observed in each biofluid and workflow. Based on each protein and peptide's analytical performance, we could describe the observable, reliable, reproducible, and quantifiable proteomes for each biofluid and workflow.
Conclusion: The standardized workflows established here allows for reproducible and quantifiable detection of proteins covering a broad dynamic range. We envisage that implementation of this standard workflow should simplify discovery approaches and facilitate the translation of candidate markers into clinical use.
Keywords: depletion; dried blood; mass spectrometry; plasma; proteomics.
© American Association for Clinical Chemistry 2021.
Conflict of interest statement
Figures
References
-
- Uzozie AC, Aebersold R. Advancing translational research and precision medicine with targeted proteomics. J Proteomics 2018;189:1–10. - PubMed
-
- Xing J, Loureiro J, Patel MT, Mikhailov D, Gusev AI. Evaluation of a novel blood microsampling device for clinical trial sample collection and protein biomarker analysis. Bioanalysis 2020;12:919–35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
