

Fortilin inhibits p53, halts cardiomyocyte apoptosis, and protects the heart against heart failure
- PMID: 34689154
- PMCID: PMC8542040
- DOI: 10.1038/s41420-021-00692-w
Fortilin inhibits p53, halts cardiomyocyte apoptosis, and protects the heart against heart failure
Abstract
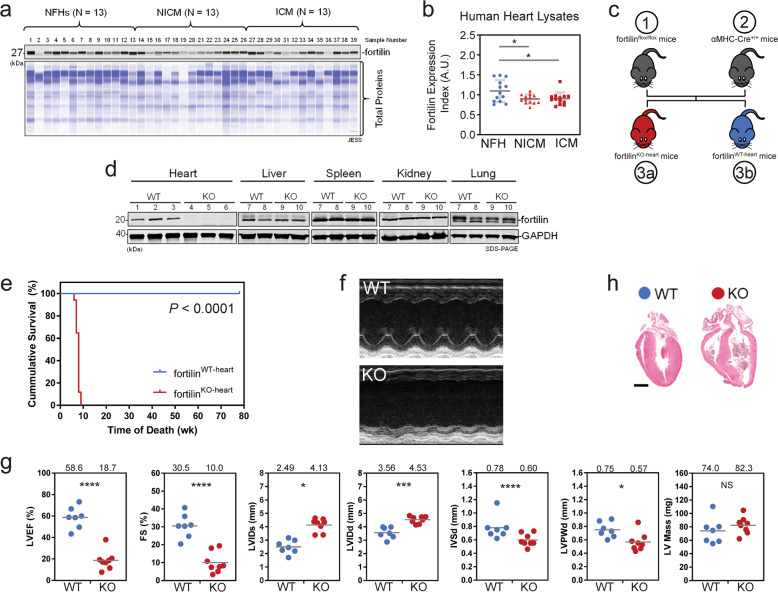
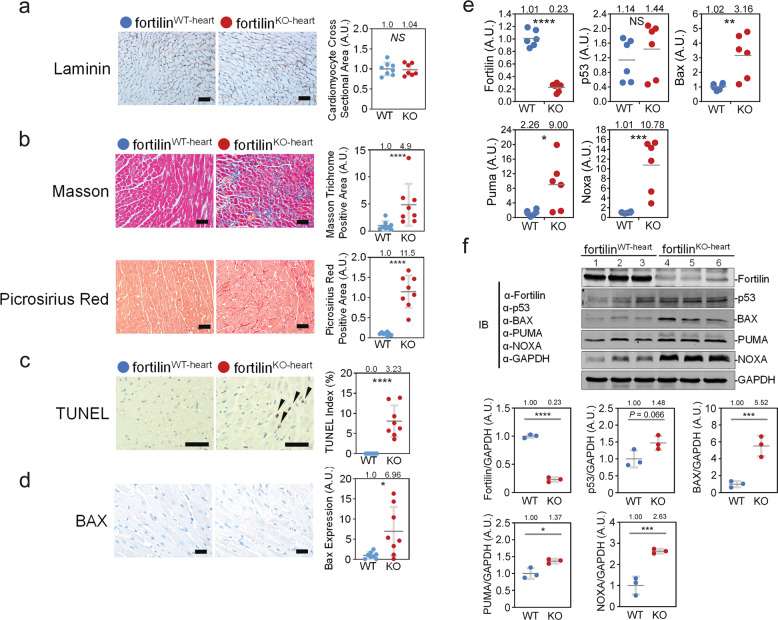
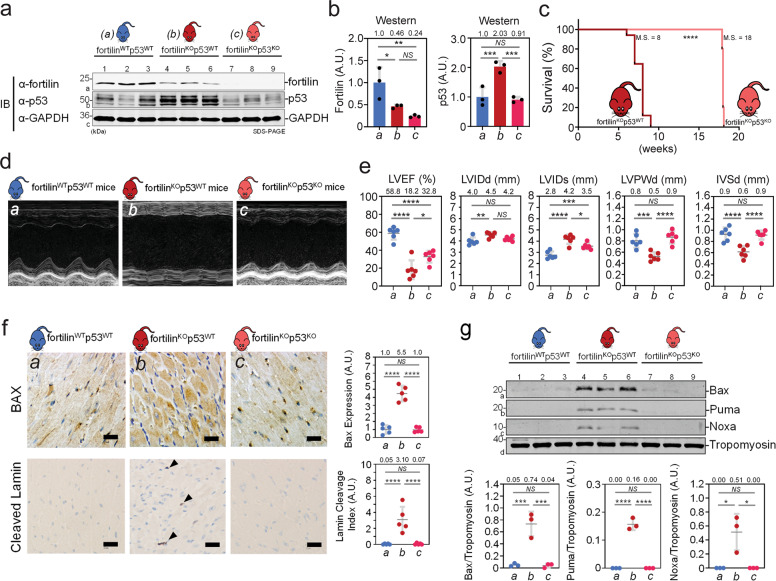
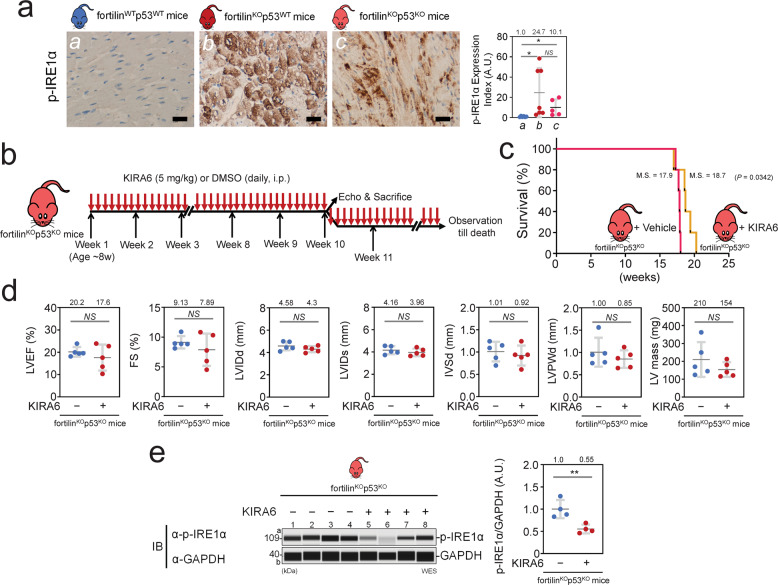
Heart failure (HF) has reached epidemic proportions in developed countries, affecting over 20 million people worldwide. Despite modern medical and device therapies, 60-70% of HF patients still die within 5 years of diagnosis as it relentlessly progresses through pervasive apoptotic loss of cardiomyocytes. Although fortilin, a 172-amino-acid anti-p53 molecule, is one of the most expressed proteins in the heart, its precise role there has remained unknown. Also unclear is how cardiomyocytes are protected against apoptosis. Here, we report that failing human hearts express less fortilin than do non-failing hearts. We also found that mice lacking fortilin in the heart (fortilinKO-heart) die by 9 weeks of age due to extensive cardiomyocyte apoptosis and severe HF, which suggests that fortilin sustains cardiomyocyte viability. The lack of fortilin is also associated with drastic upregulation of p53 target genes in the hearts. The heart-specific deletion of p53 in fortilinKO-heart mice extends their life spans from 9 to 18 weeks by mitigating cardiomyocyte apoptosis. Our data suggest that fortilin is a novel cardiac p53 inhibitor and that its inadequate expression in failing hearts and subsequent overactivation of the p53 apoptosis pathway in cardiomyocytes exacerbates HF.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Physical and functional antagonism between tumor suppressor protein p53 and fortilin, an anti-apoptotic protein.J Biol Chem. 2011 Sep 16;286(37):32575-85. doi: 10.1074/jbc.M110.217836. Epub 2011 Jul 27. J Biol Chem. 2011. PMID: 21795694 Free PMC article.
-
Fortilin potentiates the peroxidase activity of Peroxiredoxin-1 and protects against alcohol-induced liver damage in mice.Sci Rep. 2016 Jan 4;6:18701. doi: 10.1038/srep18701. Sci Rep. 2016. PMID: 26726832 Free PMC article.
-
Fortilin reduces apoptosis in macrophages and promotes atherosclerosis.Am J Physiol Heart Circ Physiol. 2013 Nov 15;305(10):H1519-29. doi: 10.1152/ajpheart.00570.2013. Epub 2013 Sep 16. Am J Physiol Heart Circ Physiol. 2013. PMID: 24043250 Free PMC article.
-
Fortilin: A Potential Target for the Prevention and Treatment of Human Diseases.Adv Clin Chem. 2017;82:265-300. doi: 10.1016/bs.acc.2017.06.006. Epub 2017 Aug 7. Adv Clin Chem. 2017. PMID: 28939212 Free PMC article. Review.
-
Anti-apoptosis in nonmyocytes and pro-autophagy in cardiomyocytes: two strategies against postinfarction heart failure through regulation of cell death/degeneration.Heart Fail Rev. 2018 Sep;23(5):759-772. doi: 10.1007/s10741-018-9708-x. Heart Fail Rev. 2018. PMID: 29737434 Review.
Cited by
-
Translationally controlled tumor protein interacts with connexin 43 and facilitates intercellular coupling between cardiomyocytes.Front Cell Dev Biol. 2025 Mar 20;13:1549063. doi: 10.3389/fcell.2025.1549063. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40181823 Free PMC article.
-
High plasma levels of fortilin are associated with cardiovascular events in patients undergoing coronary angiography.Heart Vessels. 2025 Mar;40(3):219-226. doi: 10.1007/s00380-024-02465-8. Epub 2024 Sep 28. Heart Vessels. 2025. PMID: 39342070
-
Comparative mathematical modeling reveals the differential effects of high-fat diet and ketogenic diet on the PI3K-Akt signaling pathway in heart.Nutr Metab (Lond). 2024 Aug 9;21(1):65. doi: 10.1186/s12986-024-00840-w. Nutr Metab (Lond). 2024. PMID: 39123207 Free PMC article.
-
Gastrodin Alleviates Angiotensin II-Induced Hypertension and Myocardial Apoptosis via Inhibition of the PRDX2/p53 Pathway In Vivo and In Vitro.Pharmaceuticals (Basel). 2024 Sep 12;17(9):1200. doi: 10.3390/ph17091200. Pharmaceuticals (Basel). 2024. PMID: 39338362 Free PMC article.
-
MitoQ Protects Against Oxidative Stress-Induced Mitochondrial Dysregulation in Human Cardiomyocytes.J Mol Cell Cardiol Plus. 2025 Jun 26;13:100469. doi: 10.1016/j.jmccpl.2025.100469. eCollection 2025 Sep. J Mol Cell Cardiol Plus. 2025. PMID: 40686505 Free PMC article.
References
Grants and funding
- R01 HL068024/HL/NHLBI NIH HHS/United States
- HL152723/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- HL117247/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 HL117247/HL/NHLBI NIH HHS/United States
- HL138992/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
