

Deletion of soluble epoxide hydrolase suppressed chronic kidney disease-related vascular calcification by restoring Sirtuin 3 expression
- PMID: 34689162
- PMCID: PMC8542048
- DOI: 10.1038/s41419-021-04283-6
Deletion of soluble epoxide hydrolase suppressed chronic kidney disease-related vascular calcification by restoring Sirtuin 3 expression
Abstract
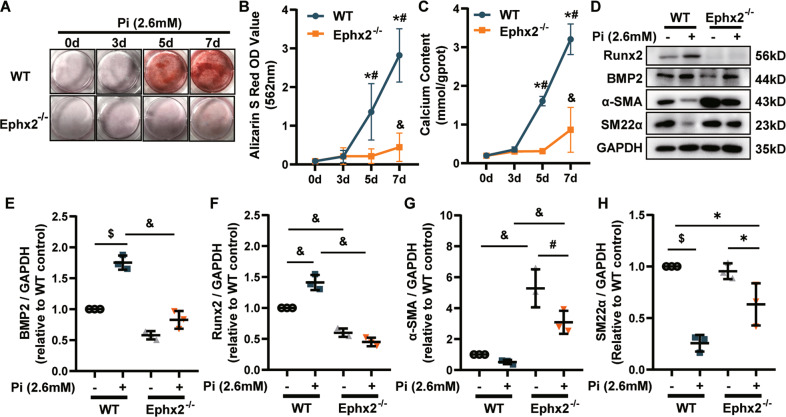
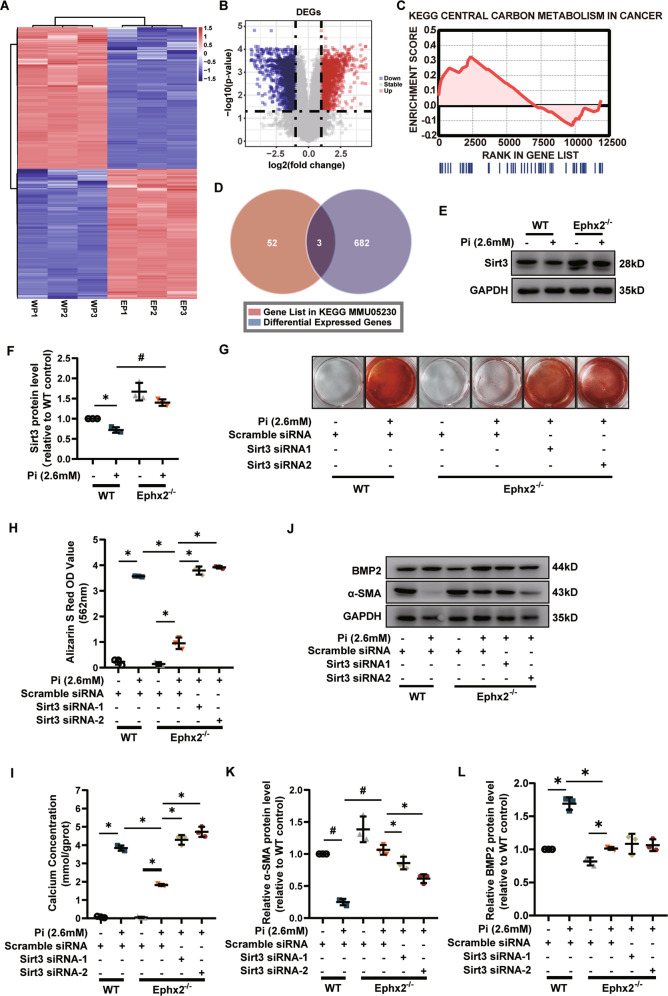
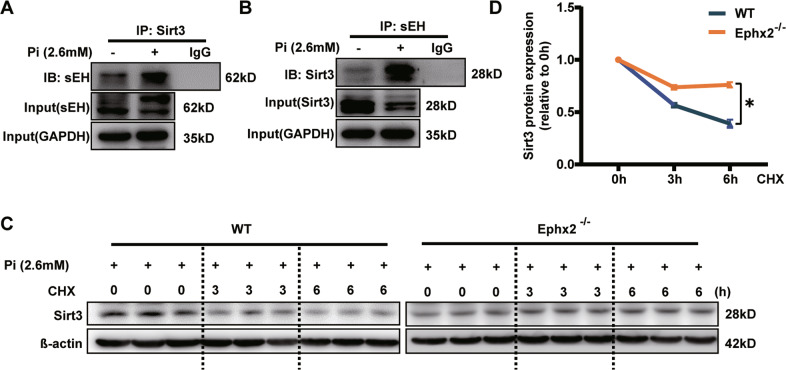
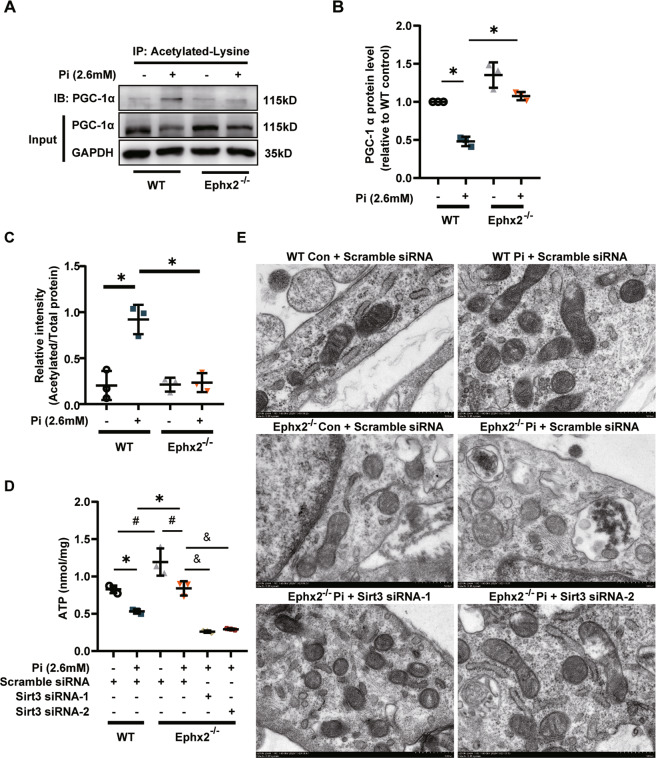
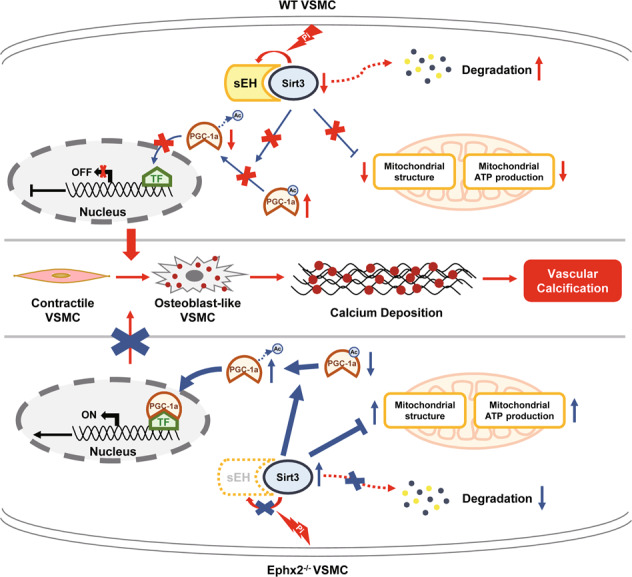
Vascular calcification is common in chronic kidney disease (CKD) and contributes to cardiovascular disease (CVD) without any effective therapies available up to date. The expression of soluble epoxide hydrolase (sEH) is different in patients with and without vascular calcification. The present study investigates the role of sEH as a potential mediator of vascular calcification in CKD. Both Ephx2-/- and wild-type (WT) mice fed with high adenine and phosphate (AP) diet were used to explore the vascular calcification in CKD. Compared with WT, deletion of sEH inhibited vascular calcification induced by AP. sEH deletion also abolished high phosphorus (Pi)-induced phenotypic transition of vascular smooth muscle cells (VSMCs) independent of its epoxyeicosatrienoic acids (EETs) hydrolysis. Further gene expression analysis identified the potential role of Sirtuin 3 (Sirt3) in the sEH-regulated VSMC calcification. Under high Pi treatment, sEH interacted with Sirt3, which might destabilize Sirt3 and accelerate the degradation of Sirt3. Deletion of sEH may preserve the expression of Sirt3, and thus maintain the mitochondrial adenosine triphosphate (ATP) synthesis and morphology, significantly suppressing VSMC calcification. Our data supported that sEH deletion inhibited vascular calcification and indicated a promising target of sEH inhibition in vascular calcification prevention.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Regulation of soluble epoxide hydrolase in renal-associated diseases: insights from potential mechanisms to clinical researches.Front Endocrinol (Lausanne). 2024 Feb 15;15:1304547. doi: 10.3389/fendo.2024.1304547. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 38425758 Free PMC article. Review.
-
Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α Inhibits Vascular Calcification Through Sirtuin 3-Mediated Reduction of Mitochondrial Oxidative Stress.Antioxid Redox Signal. 2019 Jul 1;31(1):75-91. doi: 10.1089/ars.2018.7620. Epub 2019 Apr 2. Antioxid Redox Signal. 2019. PMID: 30829051
-
Soluble epoxide hydrolase is involved in the development of atherosclerosis and arterial neointima formation by regulating smooth muscle cell migration.Am J Physiol Heart Circ Physiol. 2015 Dec 1;309(11):H1894-903. doi: 10.1152/ajpheart.00289.2015. Epub 2015 Oct 9. Am J Physiol Heart Circ Physiol. 2015. PMID: 26453326
-
Soluble epoxide hydrolase deletion attenuated nicotine-induced arterial stiffness via limiting the loss of SIRT1.Am J Physiol Heart Circ Physiol. 2021 Aug 1;321(2):H353-H368. doi: 10.1152/ajpheart.00979.2020. Epub 2021 Jun 18. Am J Physiol Heart Circ Physiol. 2021. PMID: 34142887
-
Soluble epoxide hydrolase: gene structure, expression and deletion.Gene. 2013 Sep 10;526(2):61-74. doi: 10.1016/j.gene.2013.05.008. Epub 2013 May 20. Gene. 2013. PMID: 23701967 Free PMC article. Review.
Cited by
-
Effect of C1q/TNF-Related Protein 9 on Coronary Artery Calcification: An Observational Study.J Cardiovasc Dev Dis. 2022 Sep 20;9(10):313. doi: 10.3390/jcdd9100313. J Cardiovasc Dev Dis. 2022. PMID: 36286265 Free PMC article.
-
Regulation of soluble epoxide hydrolase in renal-associated diseases: insights from potential mechanisms to clinical researches.Front Endocrinol (Lausanne). 2024 Feb 15;15:1304547. doi: 10.3389/fendo.2024.1304547. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 38425758 Free PMC article. Review.
-
Vascular, valvular and kidney calcification manifested in mouse models of adenine-induced chronic kidney disease.Ren Fail. 2023 Dec;45(1):2228920. doi: 10.1080/0886022X.2023.2228920. Ren Fail. 2023. PMID: 37369635 Free PMC article.
-
Roles of SIRT3 in aging and aging-related diseases.Int J Biol Sci. 2025 Jul 28;21(11):5135-5163. doi: 10.7150/ijbs.115518. eCollection 2025. Int J Biol Sci. 2025. PMID: 40860195 Free PMC article. Review.
-
Role of Sirtuins in Diabetes and Age-Related Processes.Cureus. 2022 Sep 4;14(9):e28774. doi: 10.7759/cureus.28774. eCollection 2022 Sep. Cureus. 2022. PMID: 36225477 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
- 82073408/National Natural Science Foundation of China (National Science Foundation of China)
- 8201101103/National Natural Science Foundation of China (National Science Foundation of China)
- 81870506/National Natural Science Foundation of China (National Science Foundation of China)
- 81422011/National Natural Science Foundation of China (National Science Foundation of China)
- 81670676/National Natural Science Foundation of China (National Science Foundation of China)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
