

PLCG1 is required for AML1-ETO leukemia stem cell self-renewal
- PMID: 34695195
- PMCID: PMC8854675
- DOI: 10.1182/blood.2021012778
PLCG1 is required for AML1-ETO leukemia stem cell self-renewal
Abstract
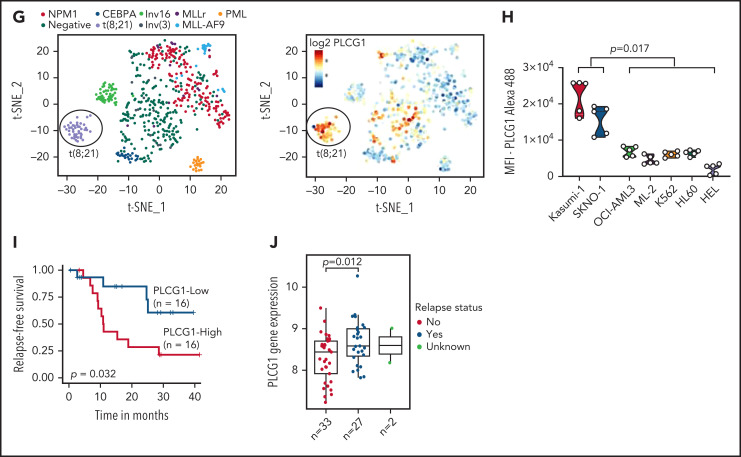
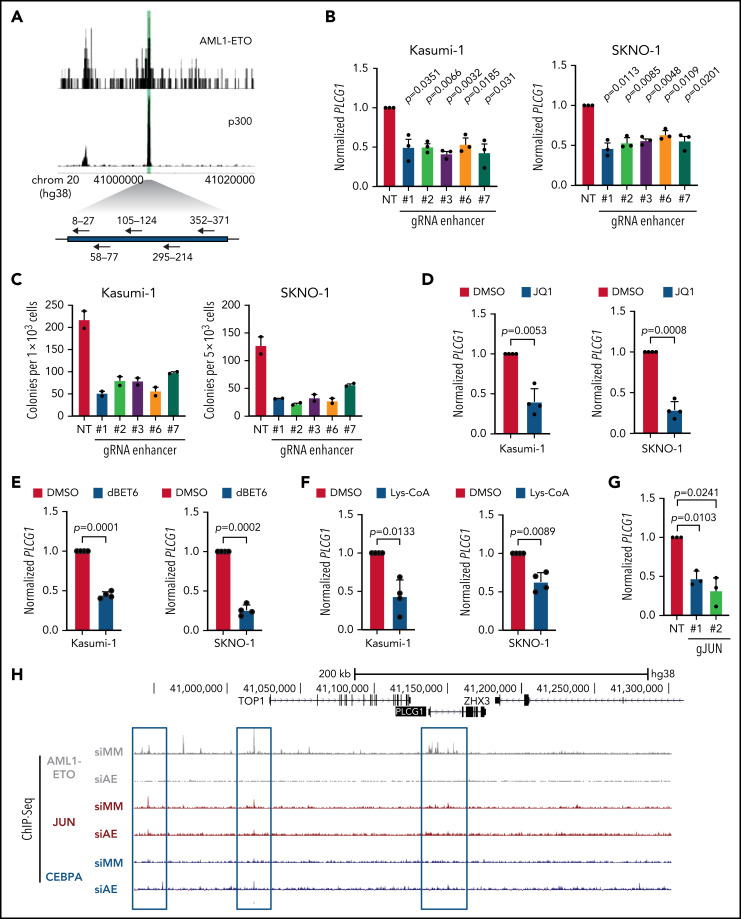
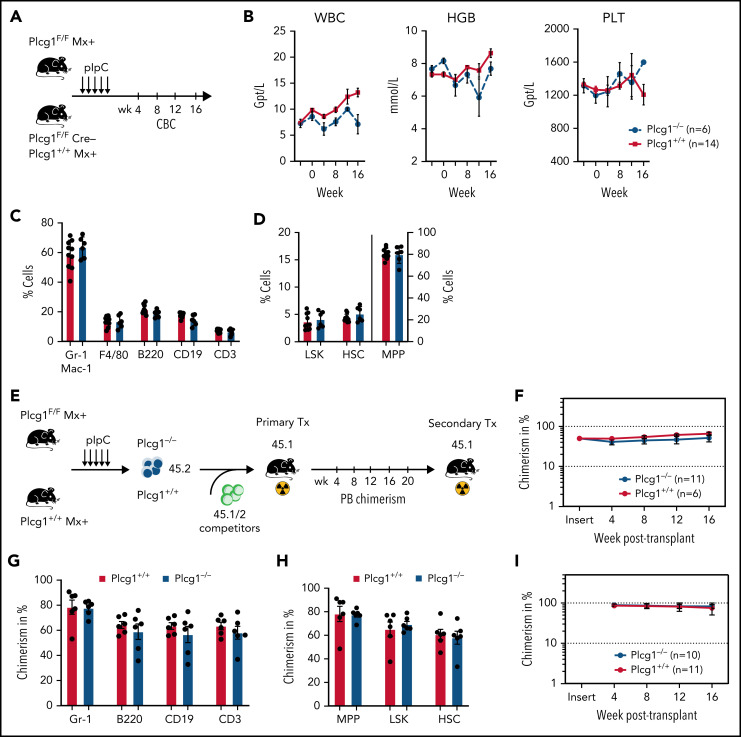
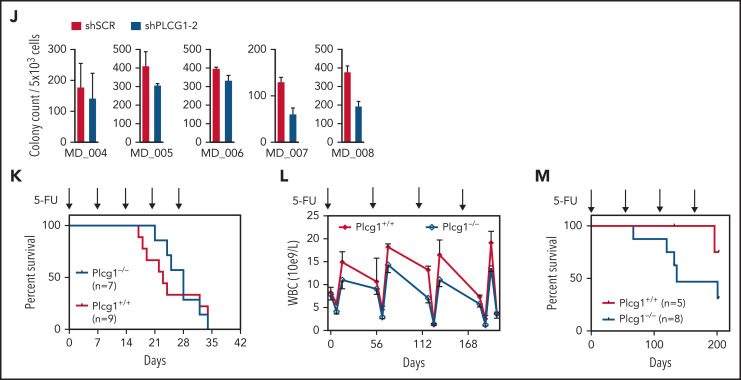
In an effort to identify novel drugs targeting fusion-oncogene-induced acute myeloid leukemia (AML), we performed high-resolution proteomic analysis. In AML1-ETO (AE)-driven AML, we uncovered a deregulation of phospholipase C (PLC) signaling. We identified PLCgamma 1 (PLCG1) as a specific target of the AE fusion protein that is induced after AE binding to intergenic regulatory DNA elements. Genetic inactivation of PLCG1 in murine and human AML inhibited AML1-ETO dependent self-renewal programs, leukemic proliferation, and leukemia maintenance in vivo. In contrast, PLCG1 was dispensable for normal hematopoietic stem and progenitor cell function. These findings are extended to and confirmed by pharmacologic perturbation of Ca++-signaling in AML1-ETO AML cells, indicating that the PLCG1 pathway poses an important therapeutic target for AML1-ETO+ leukemic stem cells.
© 2022 by The American Society of Hematology.
Figures










Comment in
-
PLaCatinG AML1-ETO.Blood. 2022 Feb 17;139(7):959-961. doi: 10.1182/blood.2021014416. Blood. 2022. PMID: 35175324 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
