

Whole-genome landscape of adult T-cell leukemia/lymphoma
- PMID: 34695199
- PMCID: PMC8854674
- DOI: 10.1182/blood.2021013568
Whole-genome landscape of adult T-cell leukemia/lymphoma
Abstract
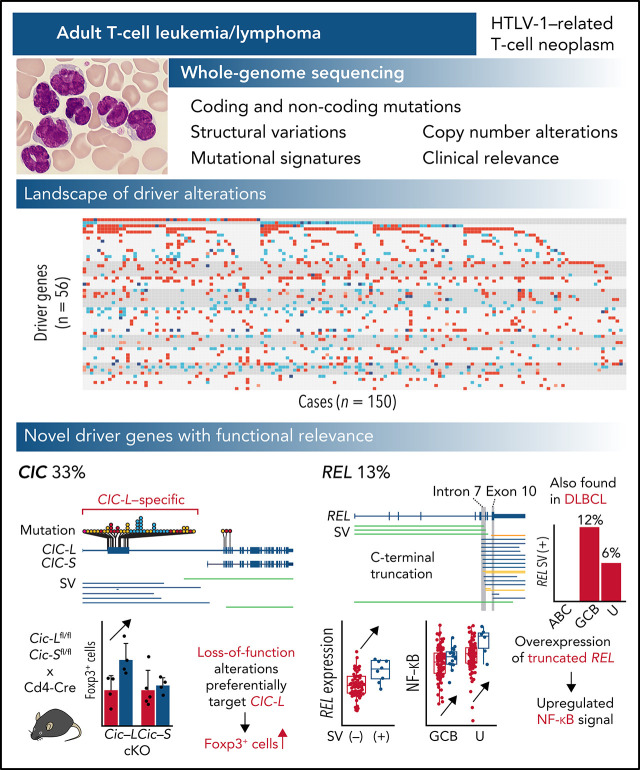
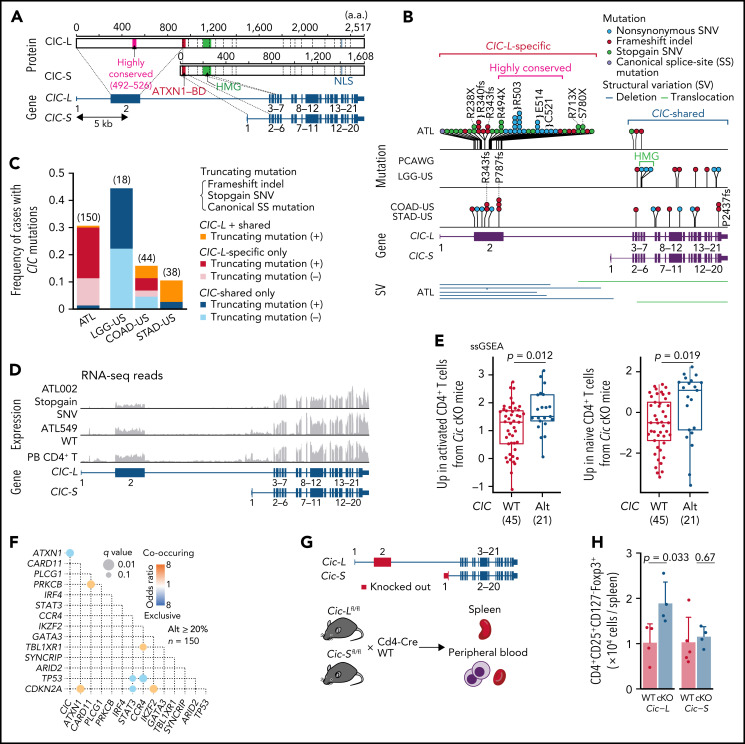
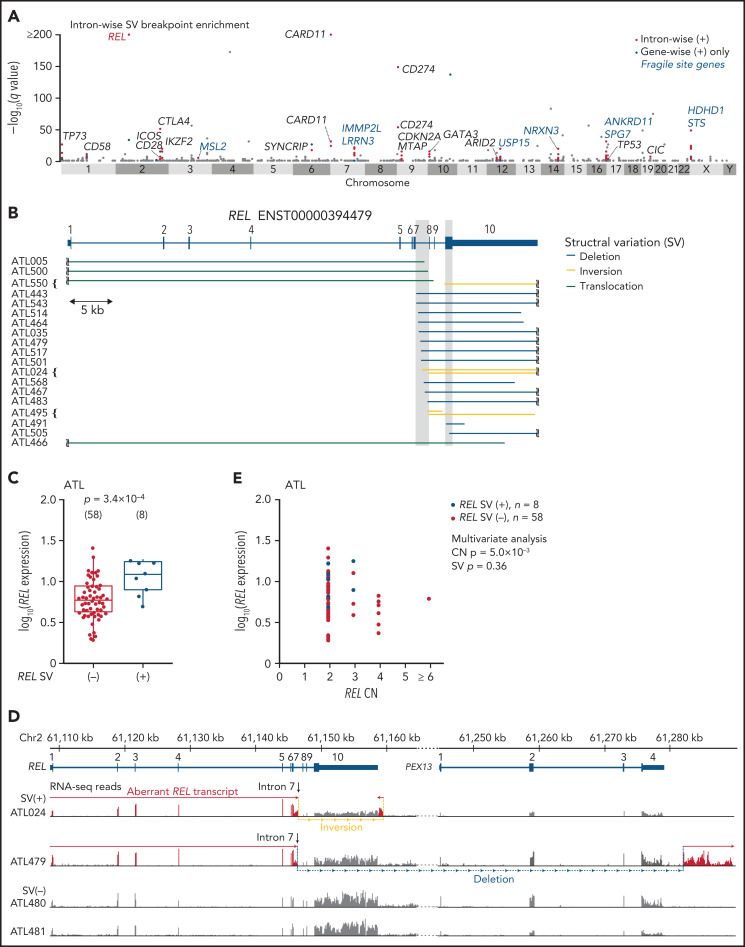
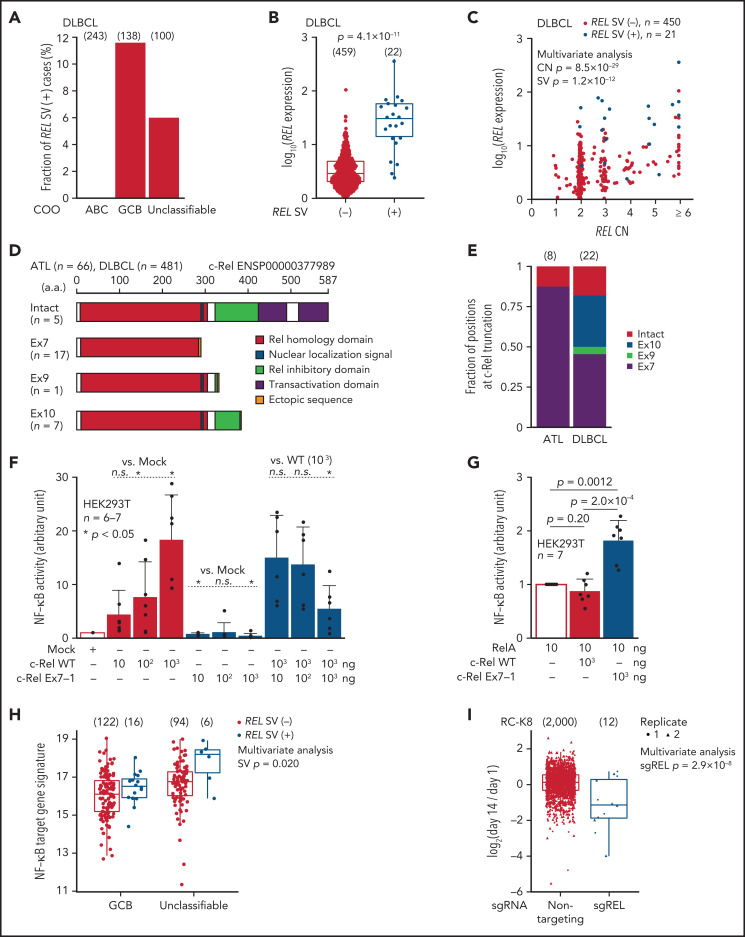
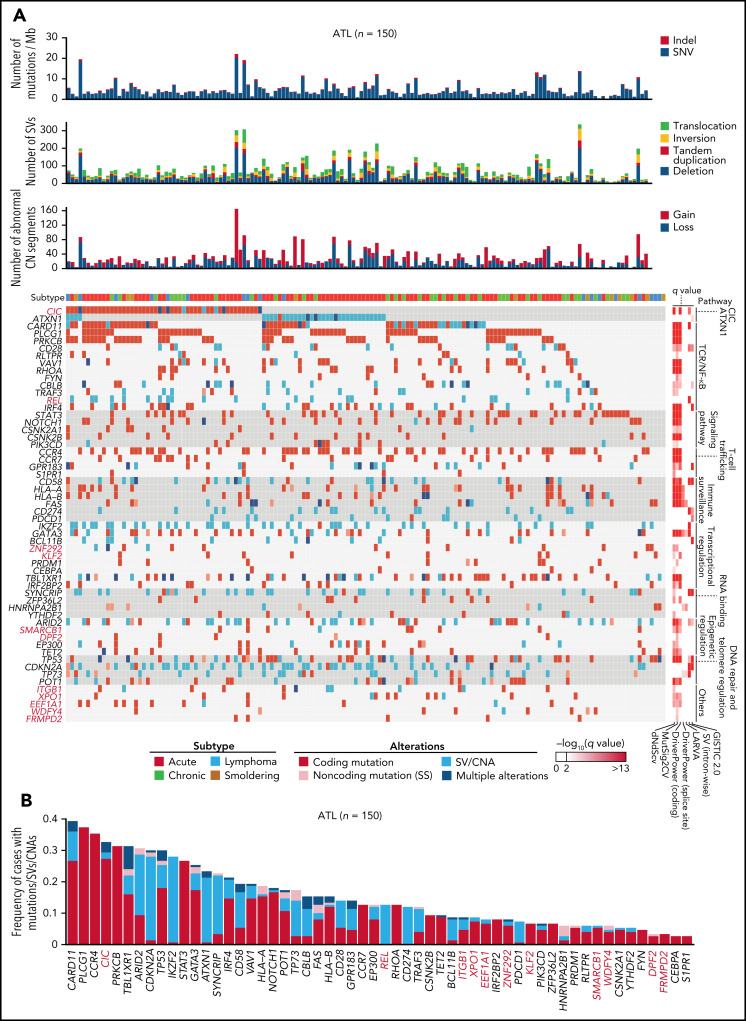
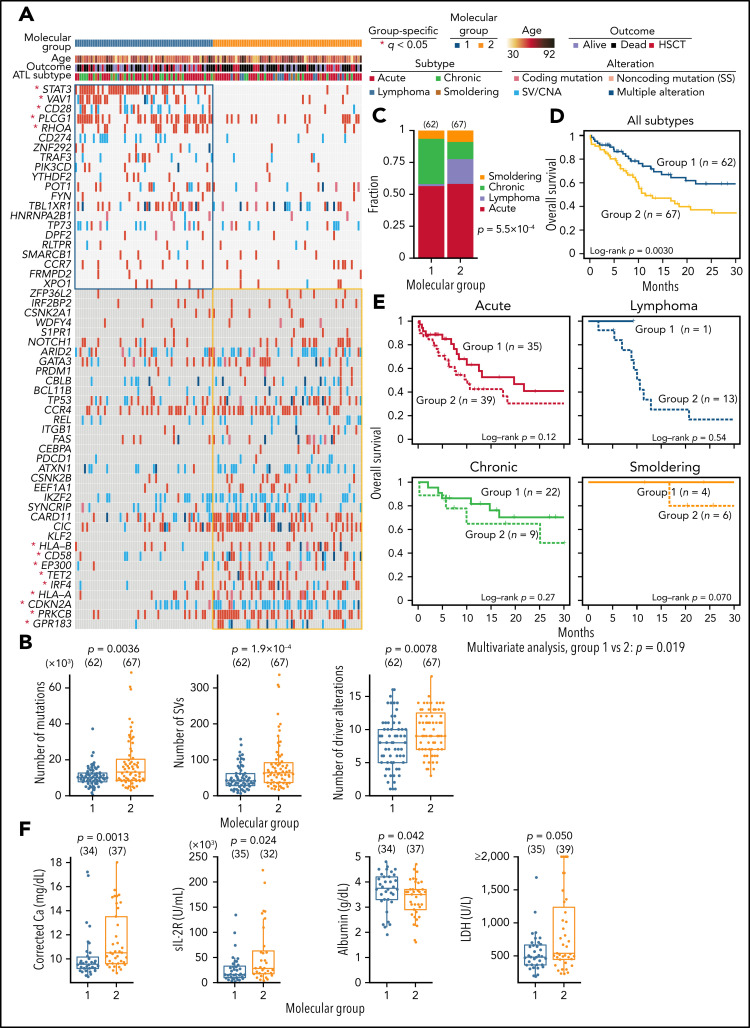
Adult T-cell leukemia/lymphoma (ATL) is an aggressive neoplasm immunophenotypically resembling regulatory T cells, associated with human T-cell leukemia virus type-1. Here, we performed whole-genome sequencing (WGS) of 150 ATL cases to reveal the overarching landscape of genetic alterations in ATL. We discovered frequent (33%) loss-of-function alterations preferentially targeting the CIC long isoform, which were overlooked by previous exome-centric studies of various cancer types. Long but not short isoform-specific inactivation of Cic selectively increased CD4+CD25+Foxp3+ T cells in vivo. We also found recurrent (13%) 3'-truncations of REL, which induce transcriptional upregulation and generate gain-of-function proteins. More importantly, REL truncations are also common in diffuse large B-cell lymphoma, especially in germinal center B-cell-like subtype (12%). In the non-coding genome, we identified recurrent mutations in regulatory elements, particularly splice sites, of several driver genes. In addition, we characterized the different mutational processes operative in clustered hypermutation sites within and outside immunoglobulin/T-cell receptor genes and identified the mutational enrichment at the binding sites of host and viral transcription factors, suggesting their activities in ATL. By combining the analyses for coding and noncoding mutations, structural variations, and copy number alterations, we discovered 56 recurrently altered driver genes, including 11 novel ones. Finally, ATL cases were classified into 2 molecular groups with distinct clinical and genetic characteristics based on the driver alteration profile. Our findings not only help to improve diagnostic and therapeutic strategies in ATL, but also provide insights into T-cell biology and have implications for genome-wide cancer driver discovery.
© 2022 by The American Society of Hematology.
Figures
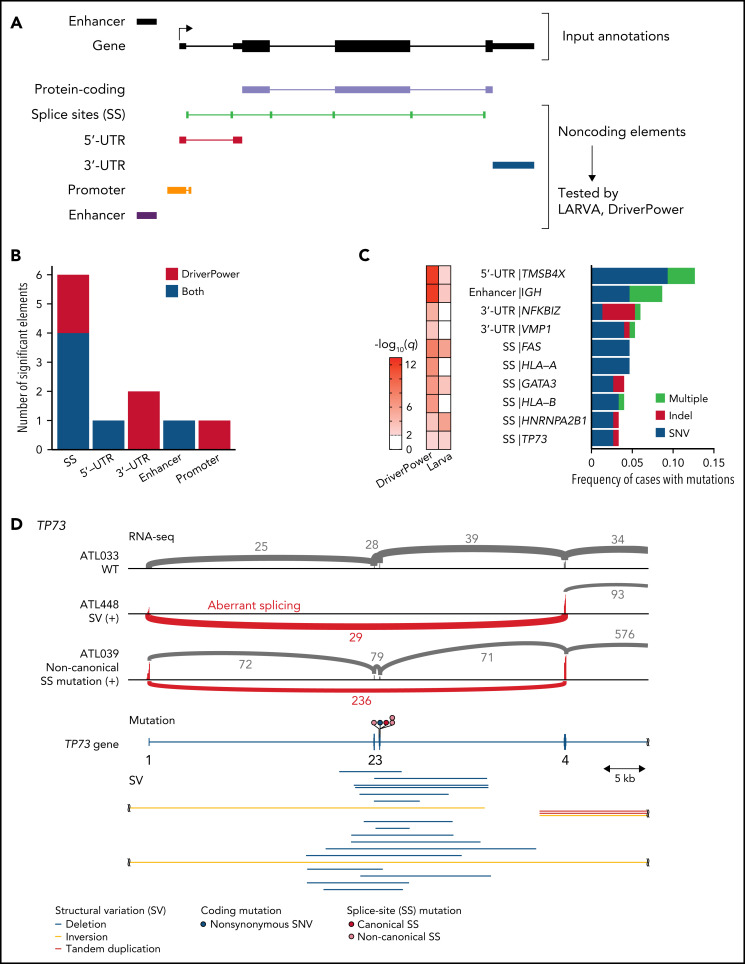
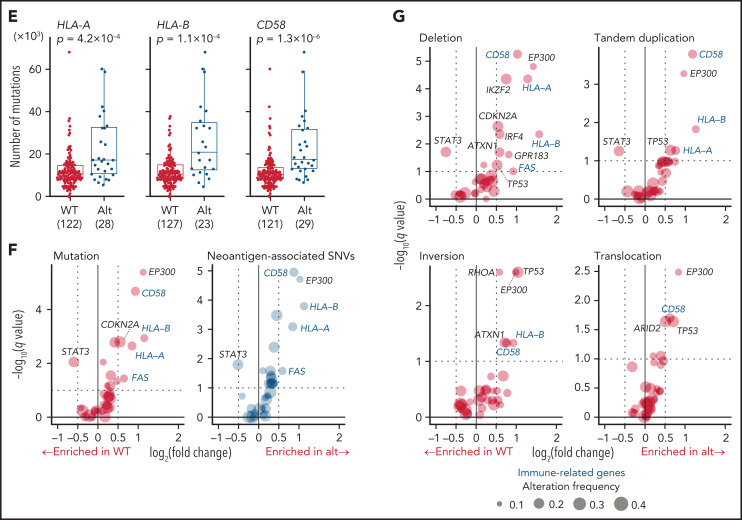
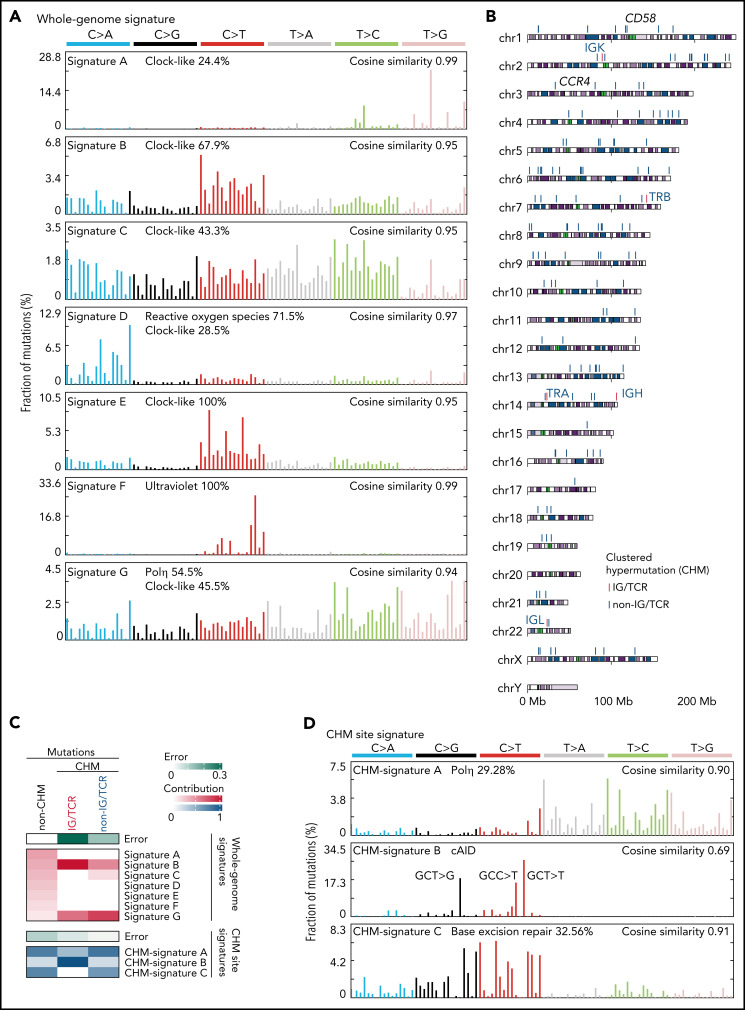
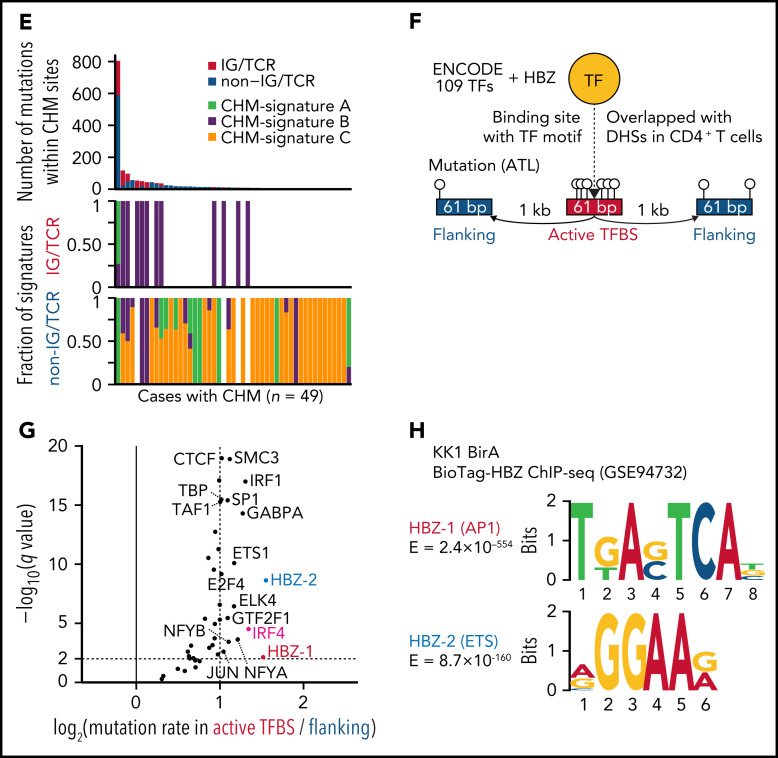
Comment in
-
Adult T-cell leukemia: genomic analysis.Blood. 2022 Feb 17;139(7):953-954. doi: 10.1182/blood.2021014450. Blood. 2022. PMID: 35175323 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
