

Mitochondrial calcium exchange in physiology and disease
- PMID: 34698550
- PMCID: PMC8816638
- DOI: 10.1152/physrev.00041.2020
Mitochondrial calcium exchange in physiology and disease
Abstract
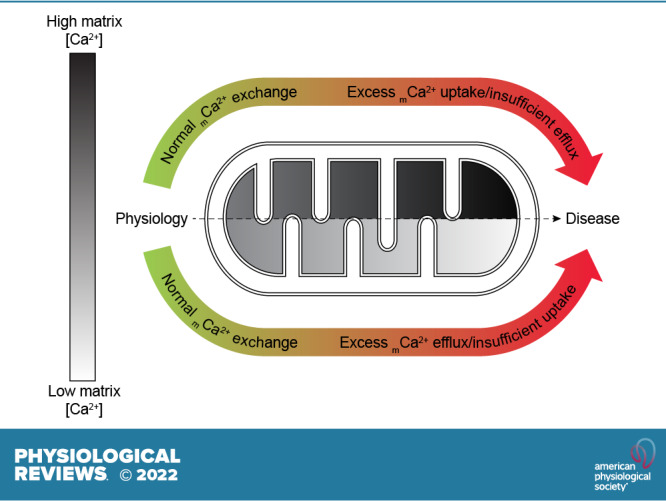
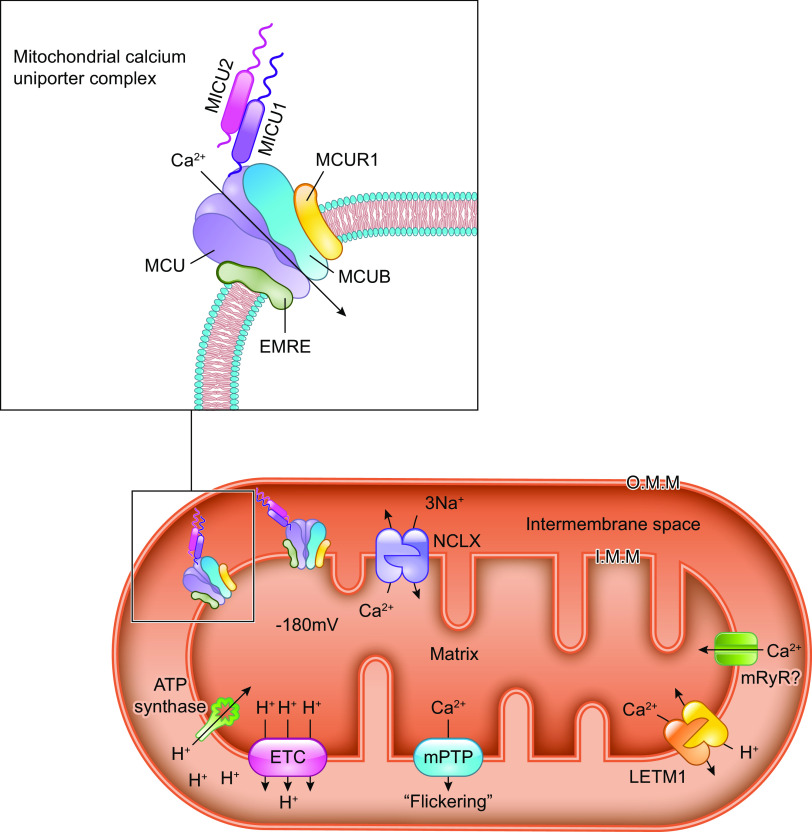
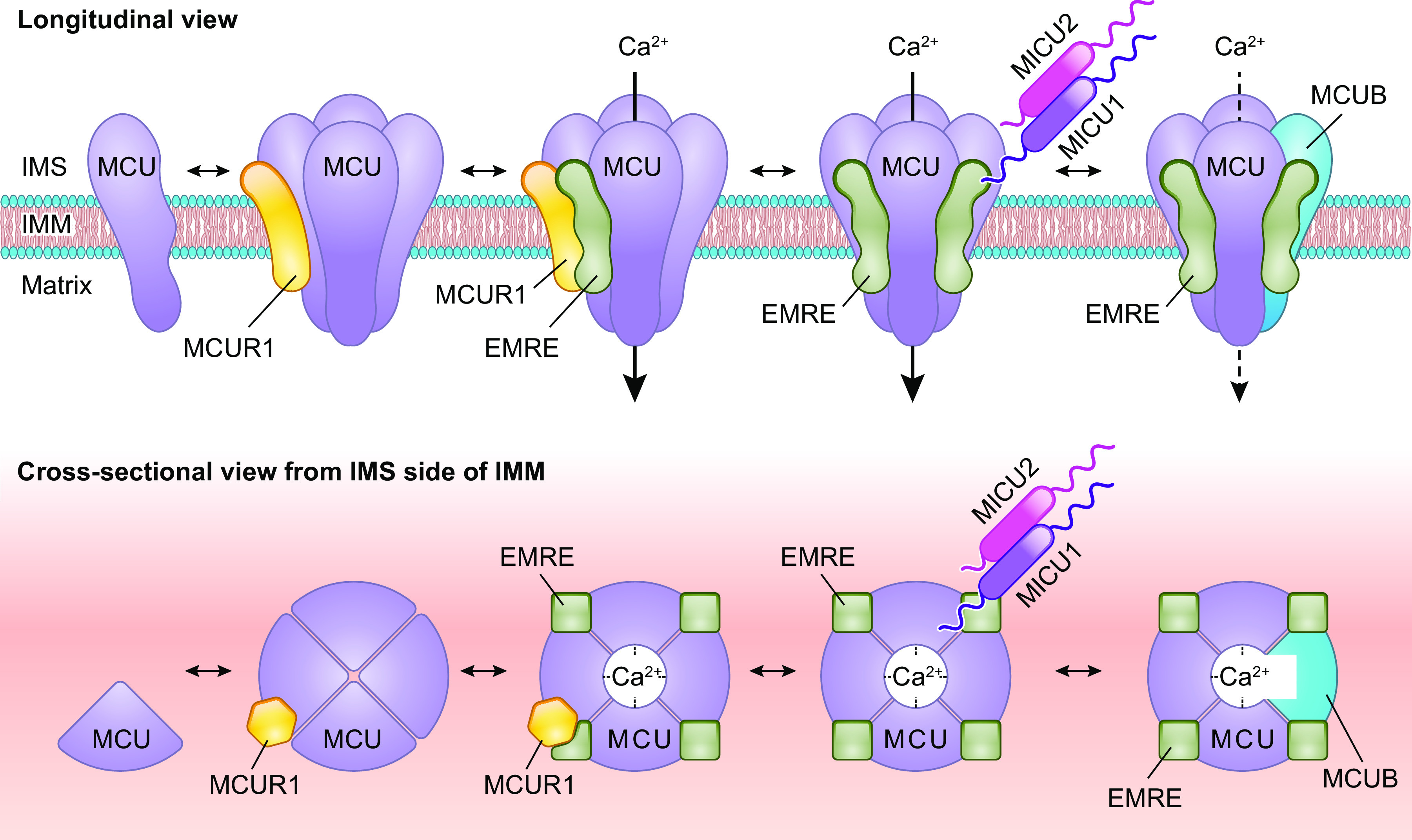
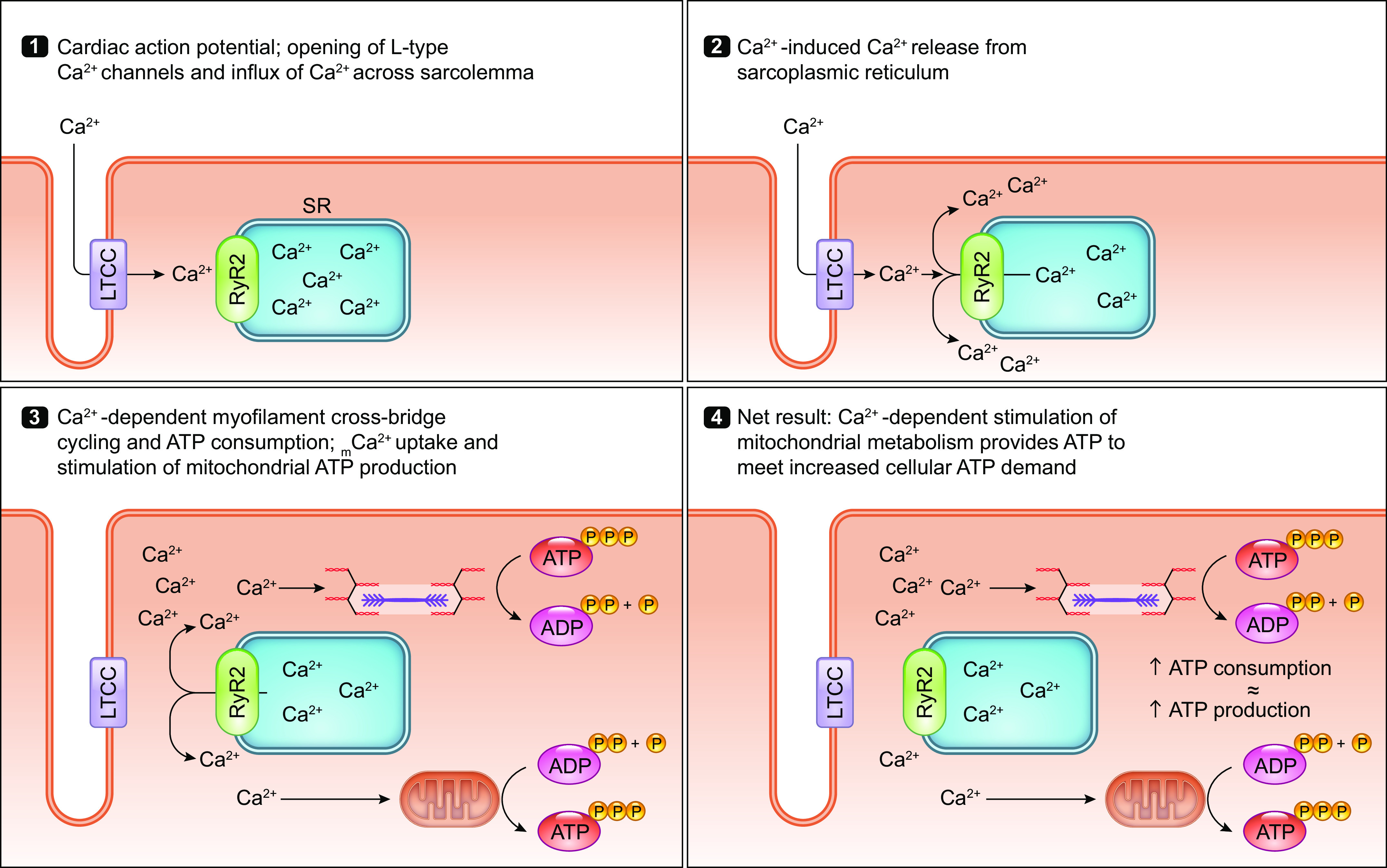
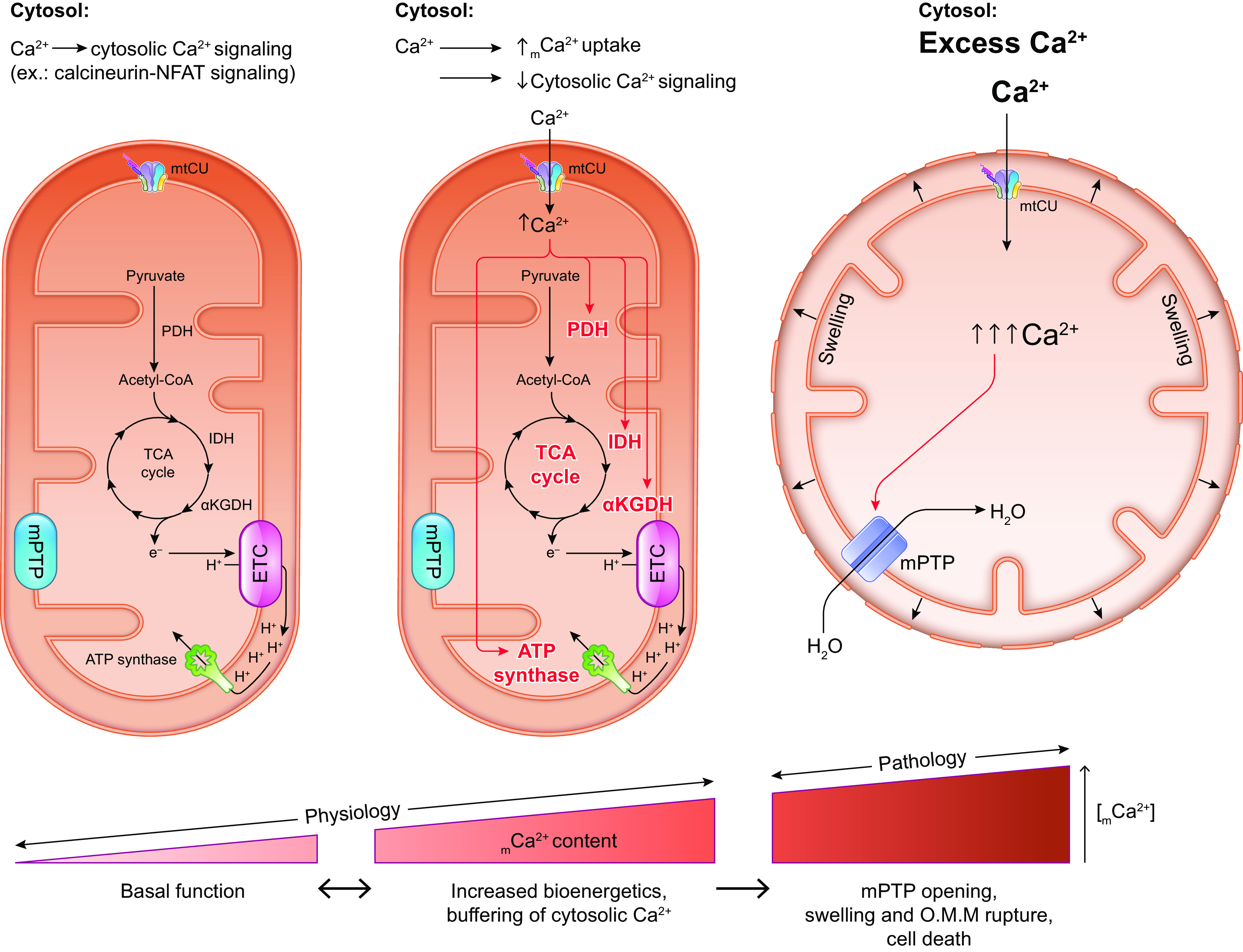
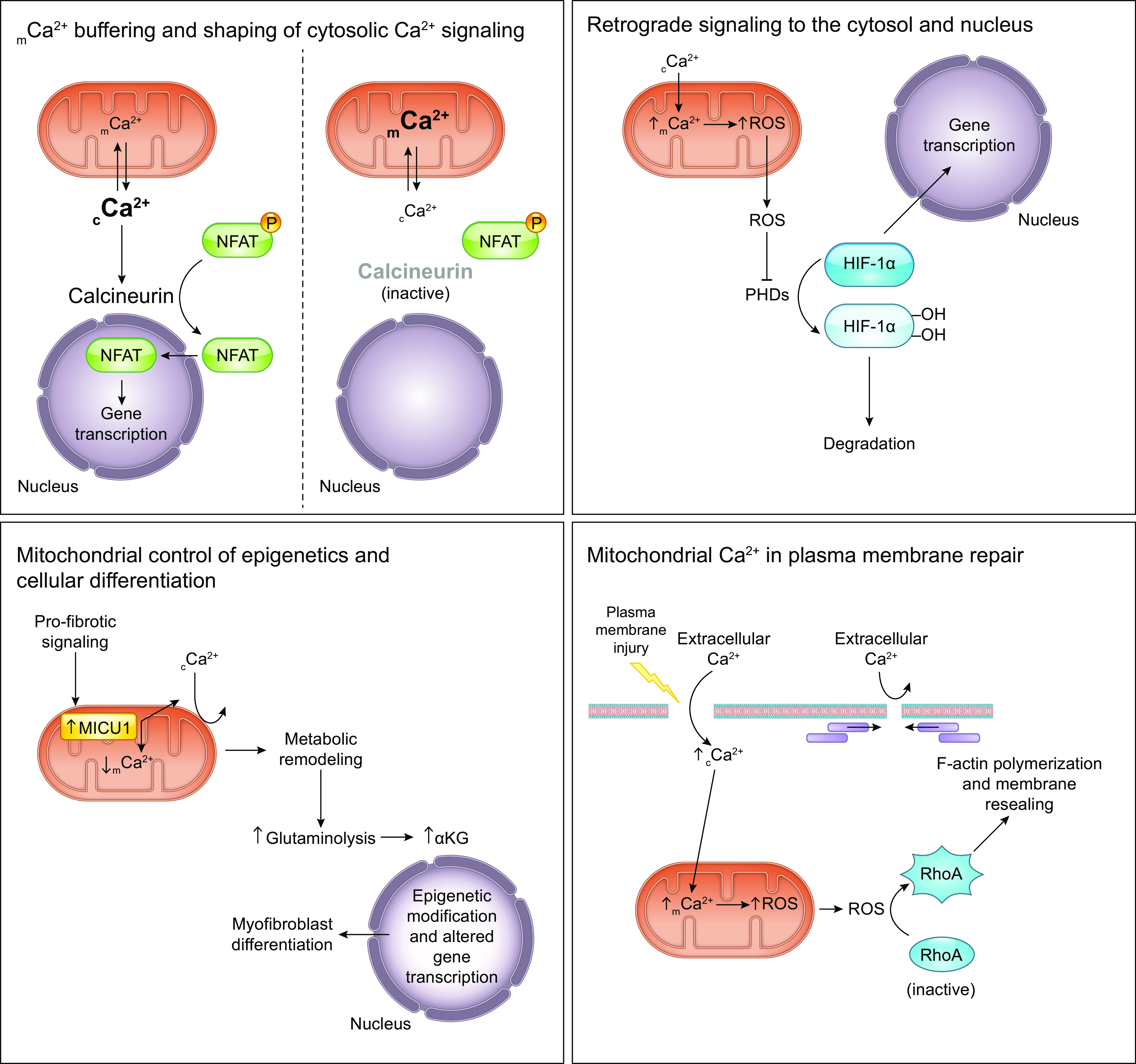
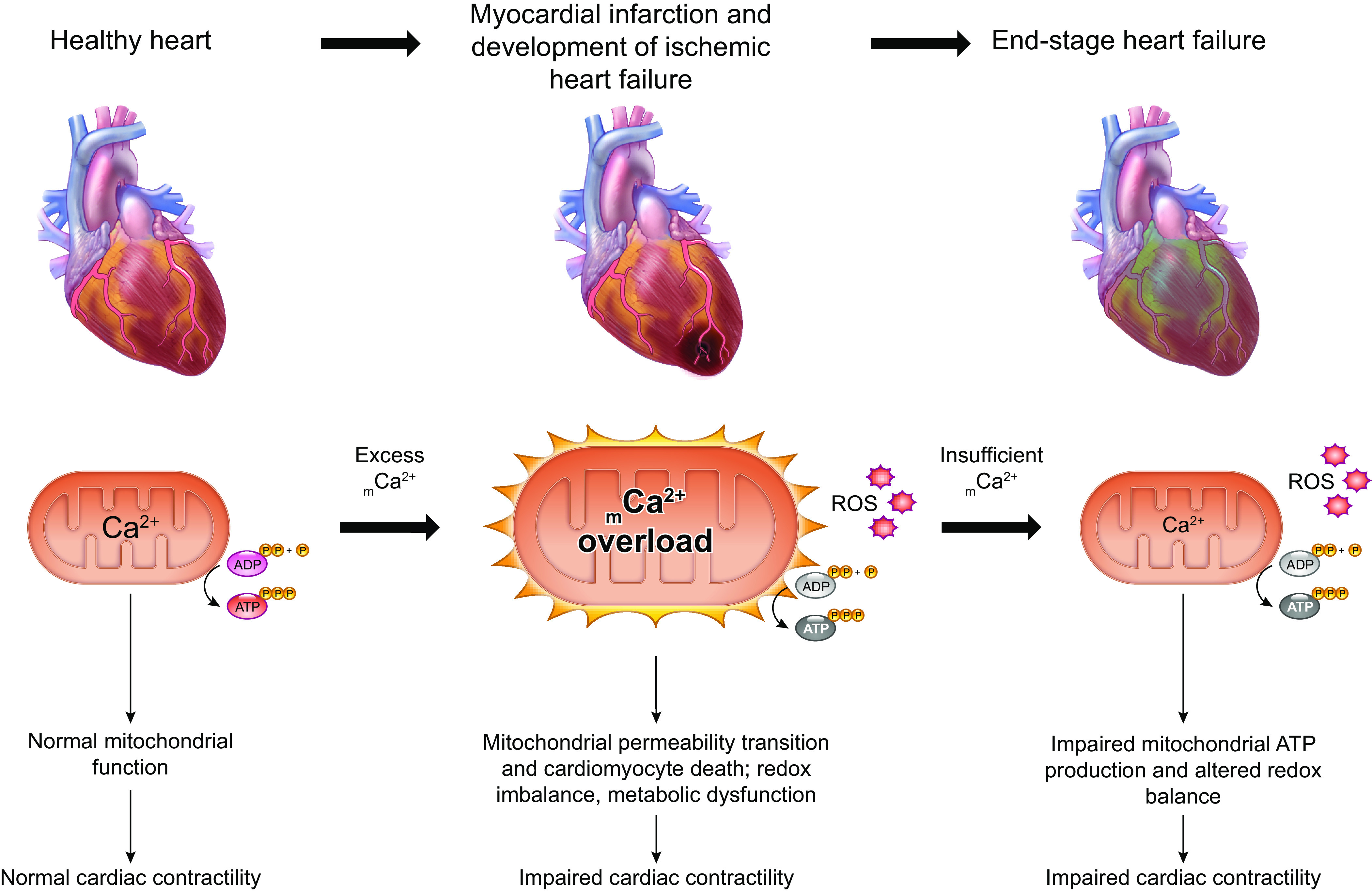
The uptake of calcium into and extrusion of calcium from the mitochondrial matrix is a fundamental biological process that has critical effects on cellular metabolism, signaling, and survival. Disruption of mitochondrial calcium (mCa2+) cycling is implicated in numerous acquired diseases such as heart failure, stroke, neurodegeneration, diabetes, and cancer and is genetically linked to several inherited neuromuscular disorders. Understanding the mechanisms responsible for mCa2+ exchange therefore holds great promise for the treatment of these diseases. The past decade has seen the genetic identification of many of the key proteins that mediate mitochondrial calcium uptake and efflux. Here, we present an overview of the phenomenon of mCa2+ transport and a comprehensive examination of the molecular machinery that mediates calcium flux across the inner mitochondrial membrane: the mitochondrial uniporter complex (consisting of MCU, EMRE, MICU1, MICU2, MICU3, MCUB, and MCUR1), NCLX, LETM1, the mitochondrial ryanodine receptor, and the mitochondrial permeability transition pore. We then consider the physiological implications of mCa2+ flux and evaluate how alterations in mCa2+ homeostasis contribute to human disease. This review concludes by highlighting opportunities and challenges for therapeutic intervention in pathologies characterized by aberrant mCa2+ handling and by summarizing critical unanswered questions regarding the biology of mCa2+ flux.
Keywords: MCU; NCLX; calcium; disease; mitochondria.
Conflict of interest statement
J.W.E. is a paid consultant for Mitobridge and Janssen. J.F.G. has no conflicts of interest, financial or otherwise, to disclose.
Figures
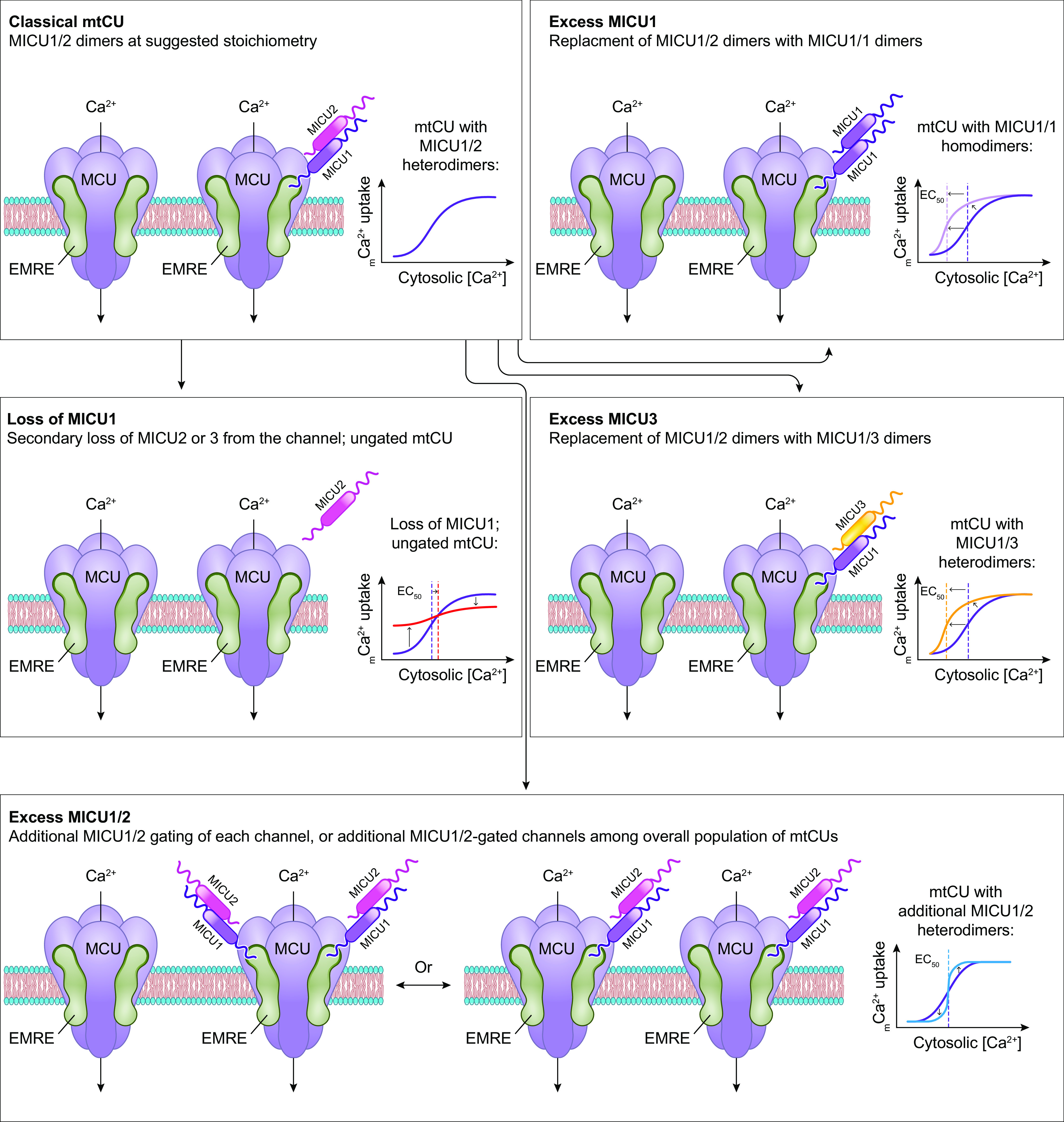
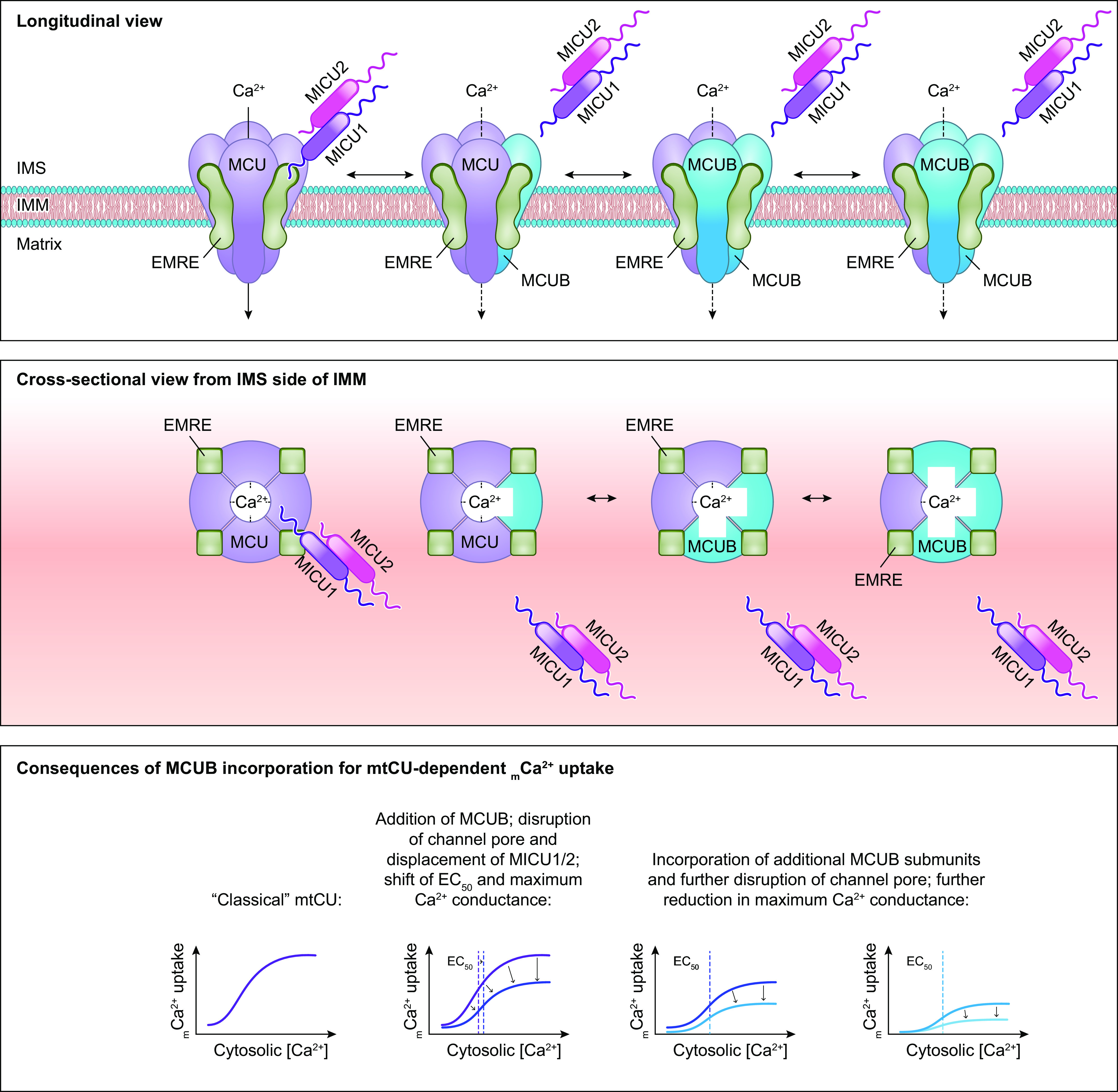
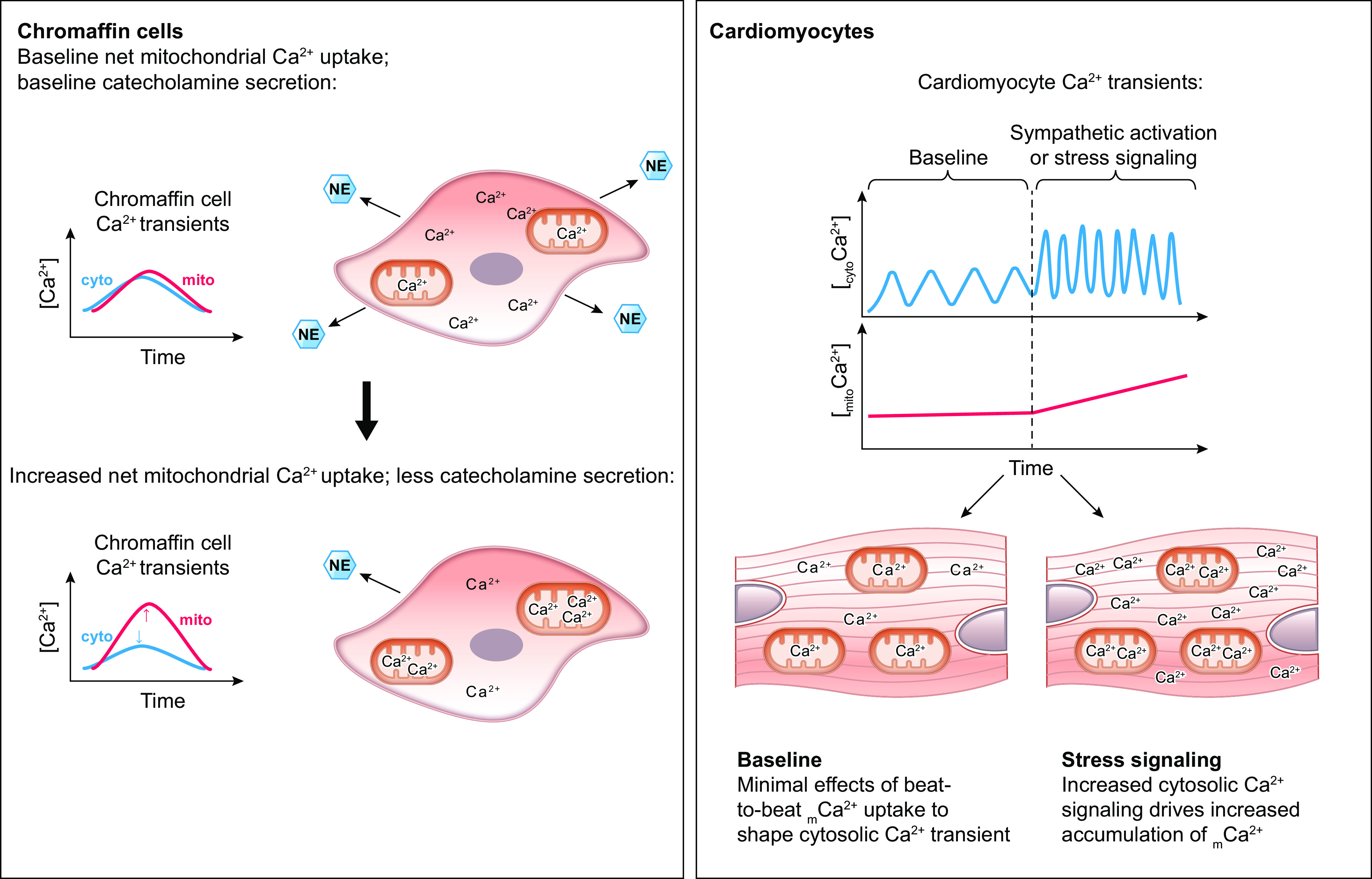
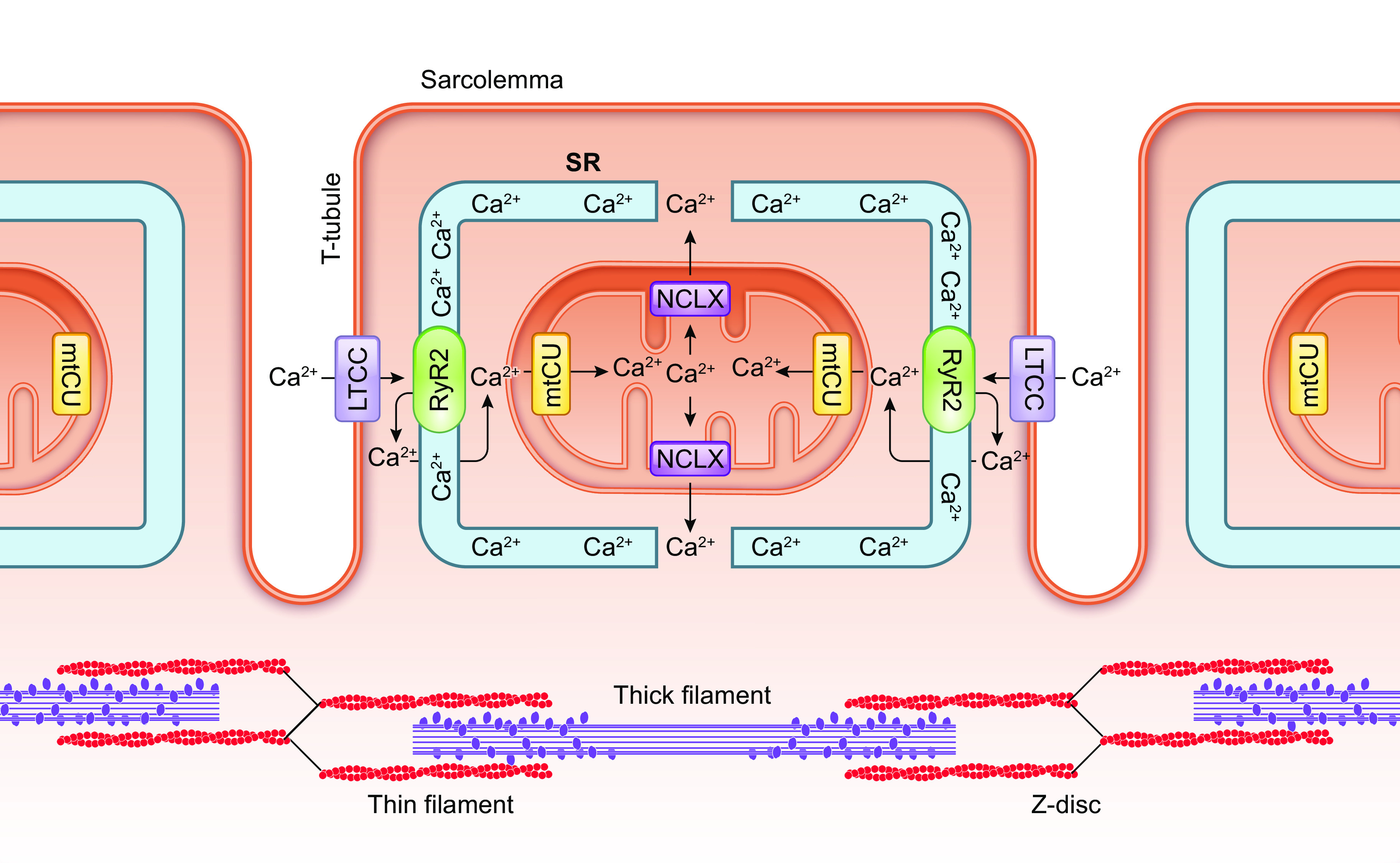
References
-
- Lehninger AL. Enzymes and Enzyme Systems: Their State in Nature. Cambridge, MA: Harvard University Press, 1951.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous