

Asxl1 loss cooperates with oncogenic Nras in mice to reprogram the immune microenvironment and drive leukemic transformation
- PMID: 34699595
- PMCID: PMC8854684
- DOI: 10.1182/blood.2021012519
Asxl1 loss cooperates with oncogenic Nras in mice to reprogram the immune microenvironment and drive leukemic transformation
Abstract
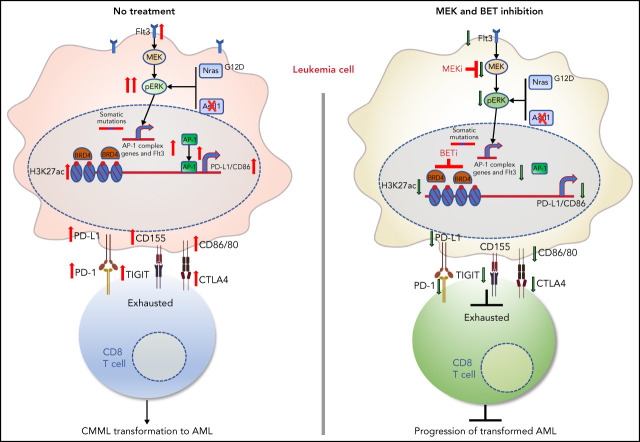
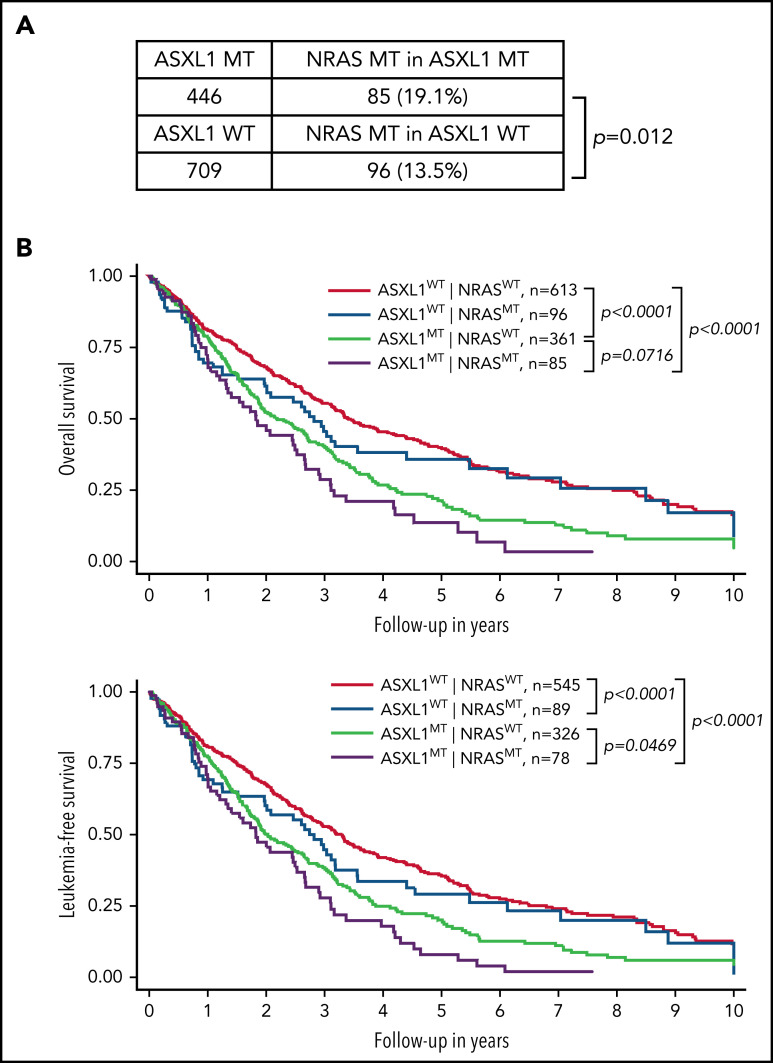
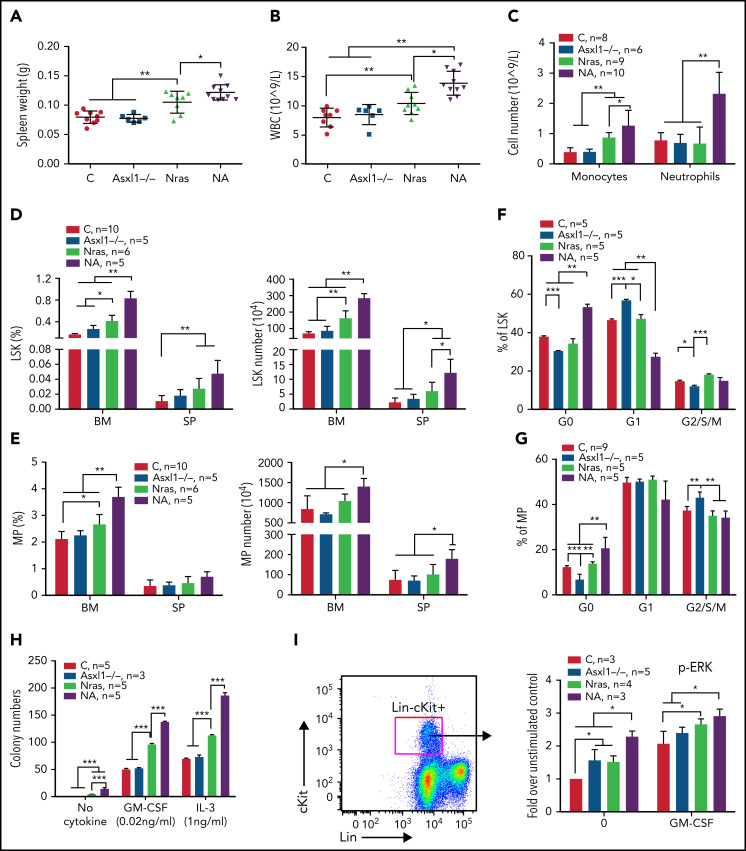
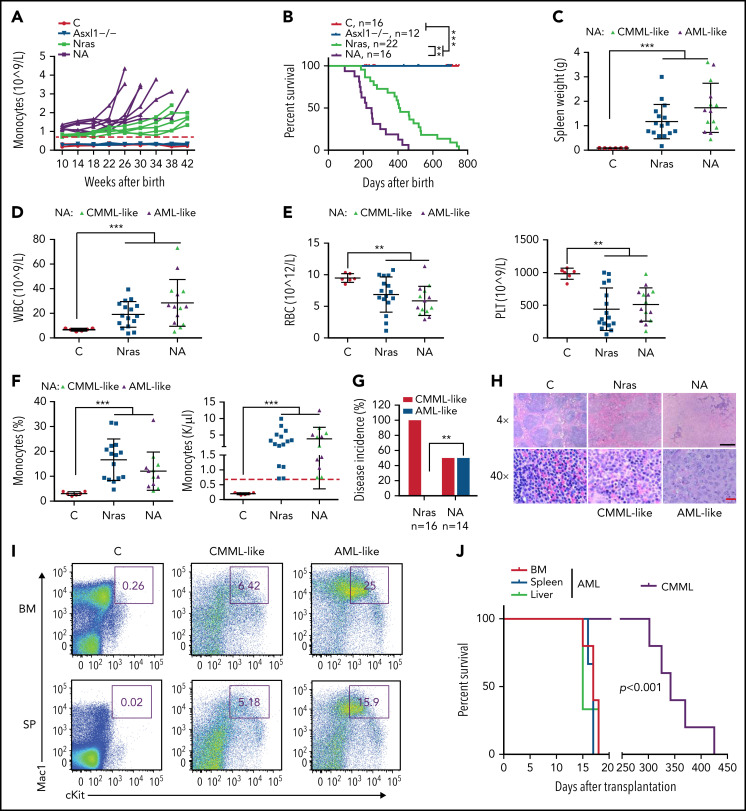
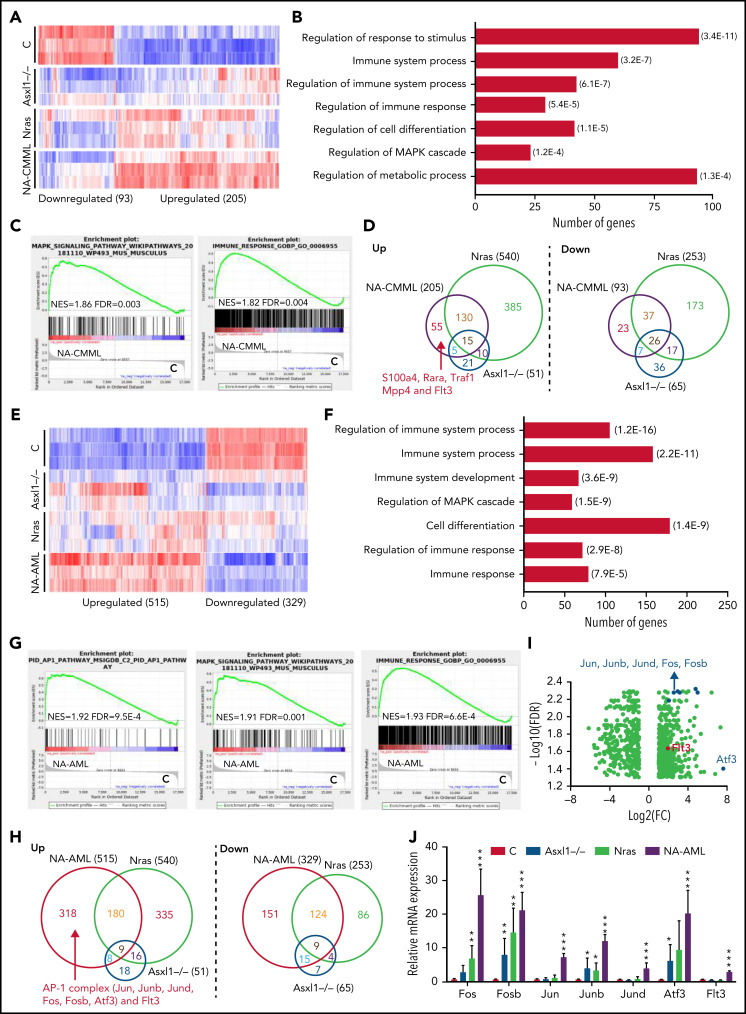
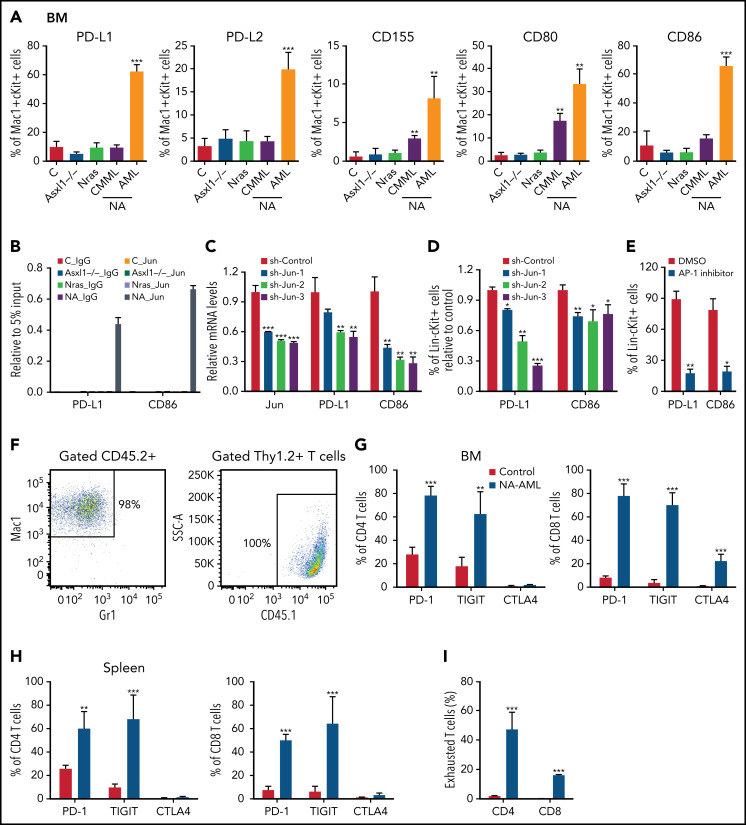
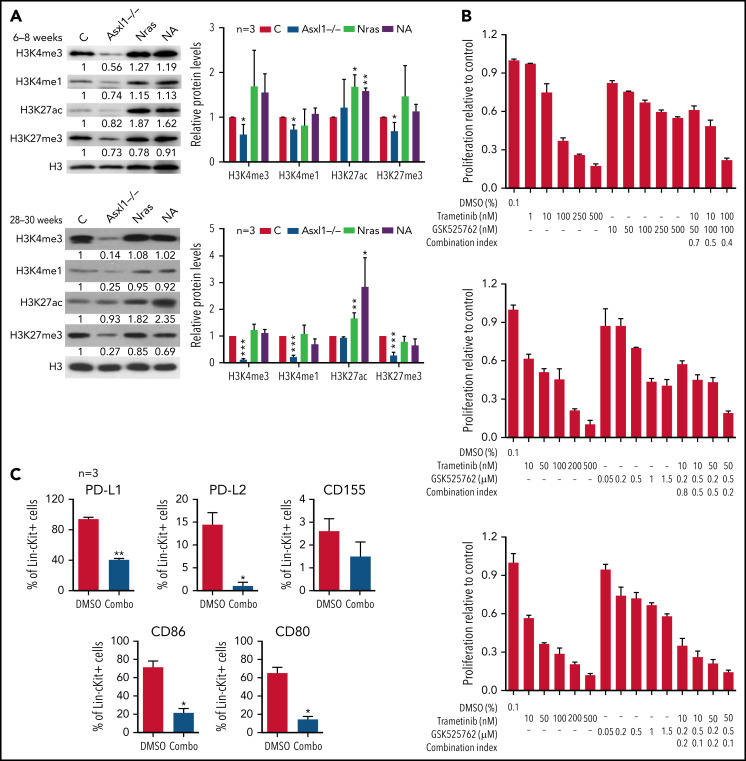
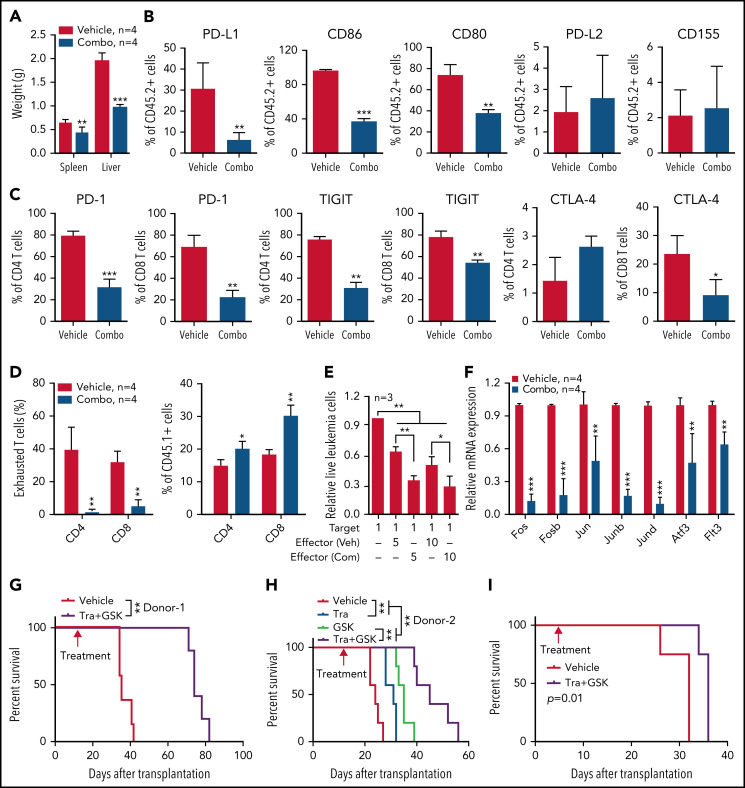
Mutations in chromatin regulator ASXL1 are frequently identified in myeloid malignancies, in particular ∼40% of patients with chronic myelomonocytic leukemia (CMML). ASXL1 mutations are associated with poor prognosis in CMML and significantly co-occur with NRAS mutations. Here, we show that concurrent ASXL1 and NRAS mutations defined a population of CMML patients who had shorter leukemia-free survival than those with ASXL1 mutation only. Corroborating this human data, Asxl1-/- accelerated CMML progression and promoted CMML transformation to acute myeloid leukemia (AML) in NrasG12D/+ mice. NrasG12D/+;Asxl1-/- (NA) leukemia cells displayed hyperactivation of MEK/ERK signaling, increased global levels of H3K27ac, upregulation of Flt3. Moreover, we find that NA-AML cells overexpressed all the major inhibitory immune checkpoint ligands: programmed death-ligand 1 (PD-L1)/PD-L2, CD155, and CD80/CD86. Among them, overexpression of PD-L1 and CD86 correlated with upregulation of AP-1 transcription factors (TFs) in NA-AML cells. An AP-1 inhibitor or short hairpin RNAs against AP-1 TF Jun decreased PD-L1 and CD86 expression in NA-AML cells. Once NA-AML cells were transplanted into syngeneic recipients, NA-derived T cells were not detectable. Host-derived wild-type T cells overexpressed programmed cell death protein 1 (PD-1) and T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT) receptors, leading to a predominant exhausted T-cell phenotype. Combined inhibition of MEK and BET resulted in downregulation of Flt3 and AP-1 expression, partial restoration of the immune microenvironment, enhancement of CD8 T-cell cytotoxicity, and prolonged survival in NA-AML mice. Our study suggests that combined targeted therapy and immunotherapy may be beneficial for treating secondary AML with concurrent ASXL1 and NRAS mutations.
© 2022 by The American Society of Hematology.
Figures
References
-
- Sinclair DA, Milne TA, Hodgson JW, et al. . The additional sex combs gene of Drosophila encodes a chromatin protein that binds to shared and unique Polycomb group sites on polytene chromosomes. Development. 1998;125(7):1207-1216. - PubMed
-
- Inoue D, Matsumoto M, Nagase R, et al. . Truncation mutants of ASXL1 observed in myeloid malignancies are expressed at detectable protein levels. Exp Hematol. 2016;44(3):172-176.e1. - PubMed
-
- Abdel-Wahab O, Dey A. The ASXL-BAP1 axis: new factors in myelopoiesis, cancer and epigenetics. Leukemia. 2013;27(1):10-15. - PubMed
-
- Inoue D, Fujino T, Sheridan P, et al. . A novel ASXL1-OGT axis plays roles in H3K4 methylation and tumor suppression in myeloid malignancies. Leukemia. 2018;32(6):1327-1337. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
