

Evaluation of two in vitro assays for tumorigenicity assessment of CRISPR-Cas9 genome-edited cells
- PMID: 34703845
- PMCID: PMC8505356
- DOI: 10.1016/j.omtm.2021.09.004
Evaluation of two in vitro assays for tumorigenicity assessment of CRISPR-Cas9 genome-edited cells
Abstract
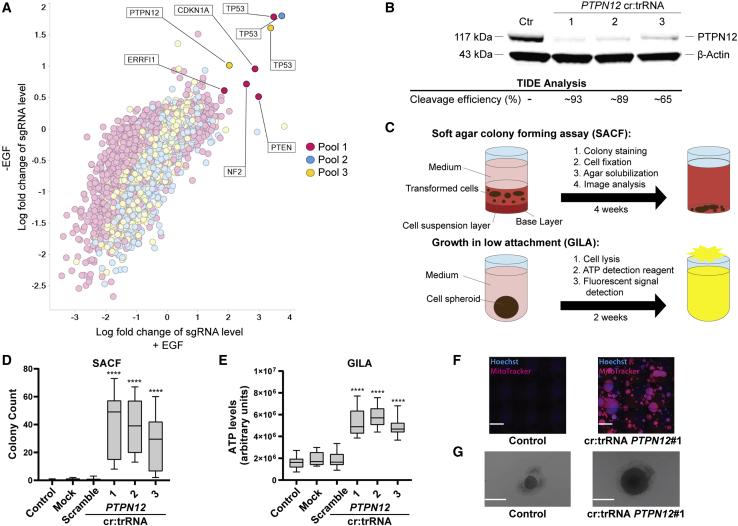
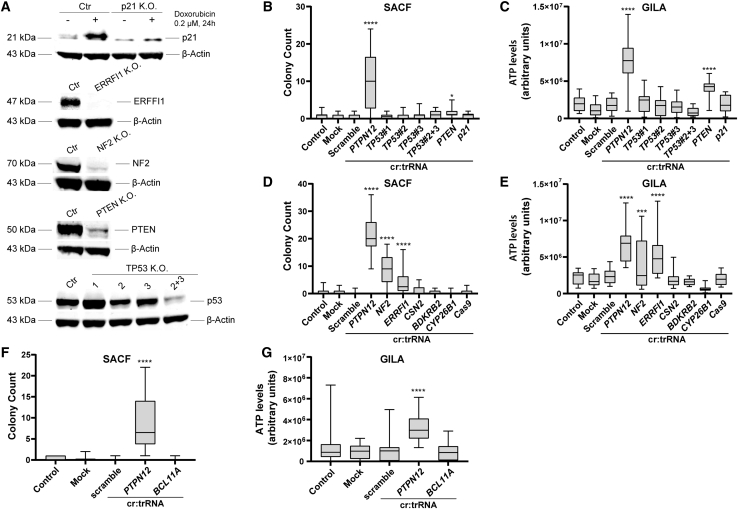
Off-target editing is one of the main safety concerns for the use of CRISPR-Cas9 genome editing in gene therapy. These unwanted modifications could lead to malignant transformation, which renders tumorigenicity assessment of gene therapy products indispensable. In this study, we established two in vitro transformation assays, the soft agar colony-forming assay (SACF) and the growth in low attachment assay (GILA) as alternative methods for tumorigenicity evaluation of genome-edited cells. Using a CRISPR-Cas9-based approach to transform immortalized MCF10A cells, we identified PTPN12, a known tumor suppressor, as a valid positive control in GILA and SACF. Next, we measured the limit of detection for both assays and proved that SACF is more sensitive than GILA (0.8% versus 3.1% transformed cells). We further validated SACF and GILA by identifying a set of positive and negative controls and by testing the suitability of another cell line (THLE-2). Moreover, in contrast to SACF and GILA, an in vivo tumorigenicity study failed to detect the known tumorigenic potential of PTPN12 deletion, demonstrating the relevance of GILA and SACF in tumorigenicity testing. In conclusion, SACF and GILA are both attractive and valuable additions to preclinical safety assessment of gene therapy products.
Keywords: 3Rs; CRISPR-Cas9; anchorage independency; gene therapy; genome editing; in vitro transformation assay; preclinical safety; soft agar.
© 2021 The Authors.
Conflict of interest statement
M.L., B.F., M.Z., L.R., G.K., A.d.R.-E., F.S., D.M., V.D., A.P., A.M., U.P.-H., U.N., J.H., H.-J.M., and S.L. were employees of the Novartis Institutes for Biomedical Research at the time this research was conducted. A.O. declares no competing interests.
Figures



References
LinkOut - more resources
Full Text Sources
