

Flexible CDOCKER: Hybrid Searching Algorithm and Scoring Function with Side Chain Conformational Entropy
- PMID: 34704754
- PMCID: PMC8684595
- DOI: 10.1021/acs.jcim.1c01078
Flexible CDOCKER: Hybrid Searching Algorithm and Scoring Function with Side Chain Conformational Entropy
Abstract
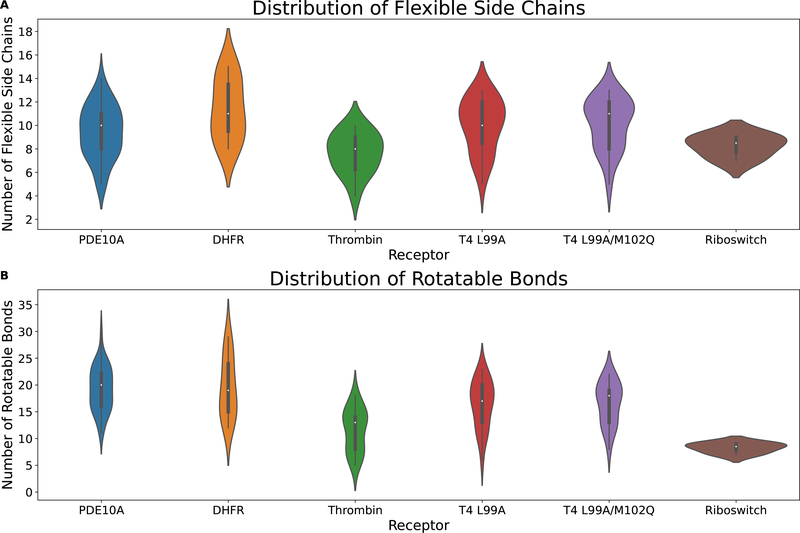
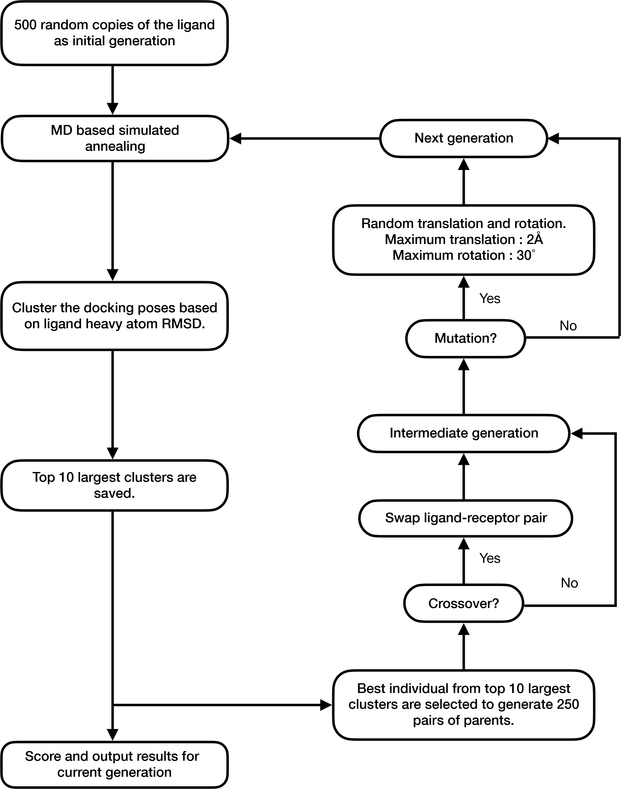
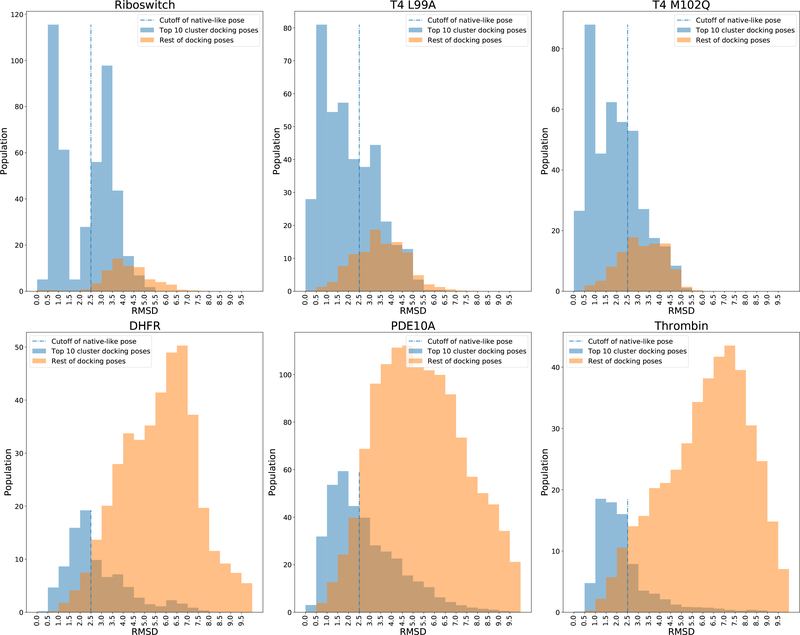
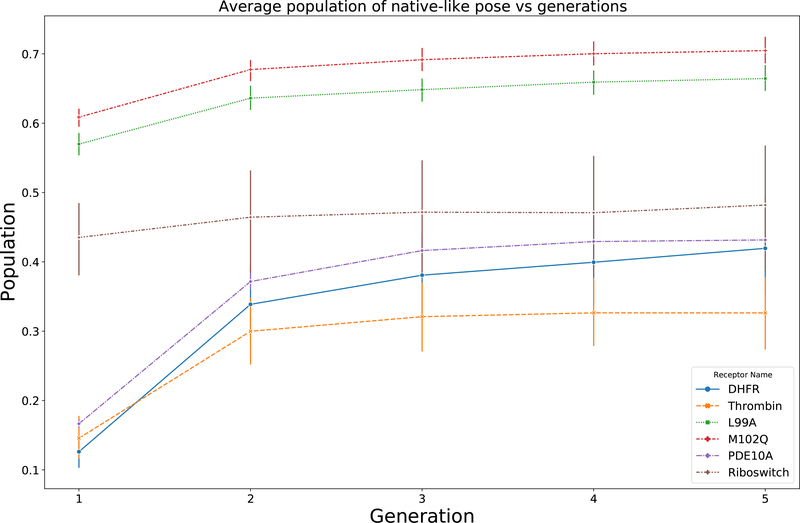
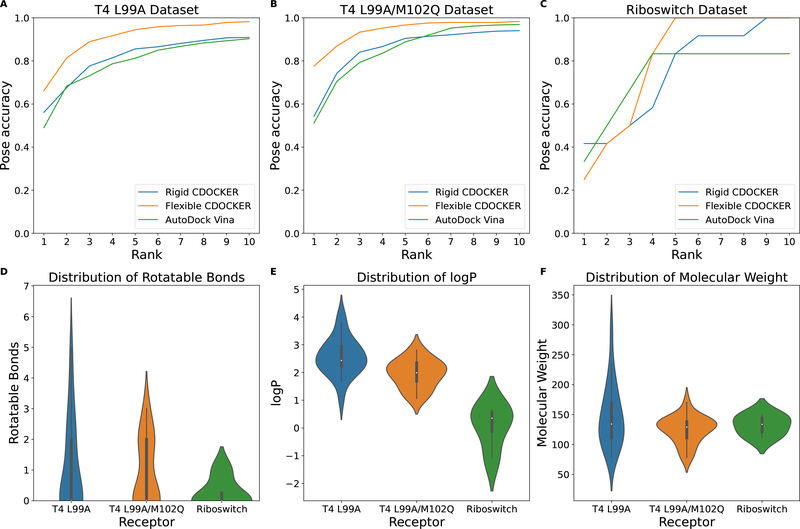
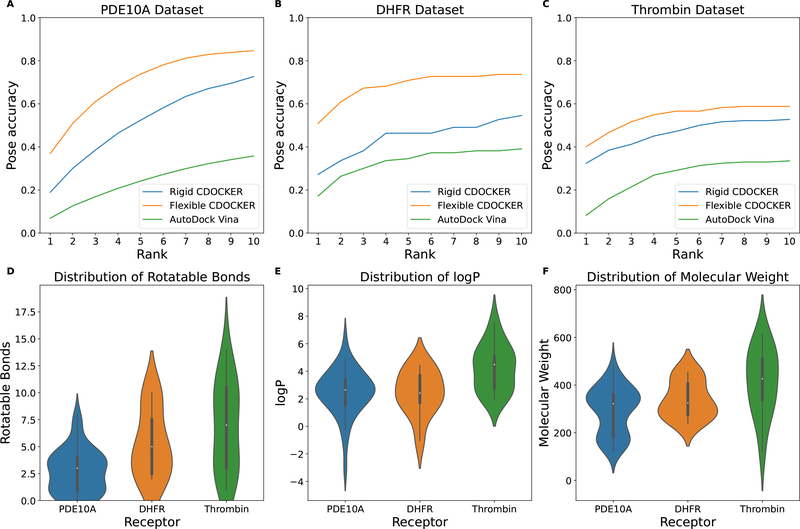
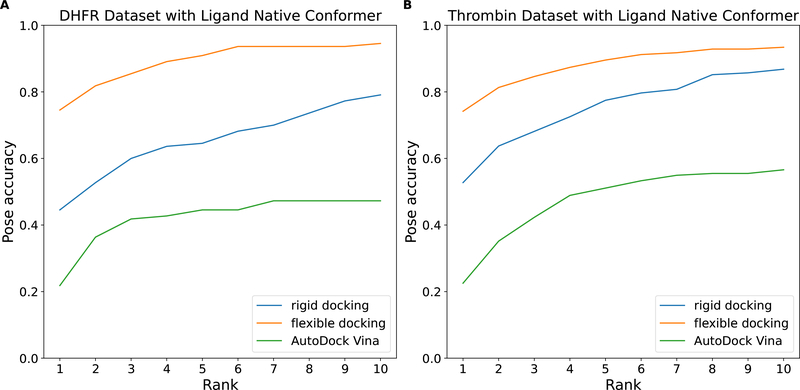
The binding of small-molecule ligands to protein or nucleic acid targets is important to numerous biological processes. Accurate prediction of the binding modes between a ligand and a macromolecule is of fundamental importance in structure-based structure-function exploration. When multiple ligands with different sizes are docked to a target receptor, it is reasonable to assume that the residues in the binding pocket may adopt alternative conformations upon interacting with the different ligands. In addition, it has been suggested that the entropic contribution to binding can be important. However, only a few attempts to include the side chain conformational entropy upon binding within the application of flexible receptor docking methodology exist. Here, we propose a new physics-based scoring function that includes both enthalpic and entropic contributions upon binding by considering the conformational variability of the flexible side chains within the ensemble of docked poses. We also describe a novel hybrid searching algorithm that combines both molecular dynamics (MD)-based simulated annealing and genetic algorithm crossovers to address the enhanced sampling of the increased search space. We demonstrate improved accuracy in flexible cross-docking experiments compared with rigid cross-docking. We test our developments by considering five protein targets, thrombin, dihydrofolate reductase(DHFR), T4 L99A, T4 L99A/M102Q, and PDE10A, which belong to different enzyme classes with different binding pocket environments, as a representative set of diverse ligands and receptors. Each target contains dozens of different ligands bound to the same binding pocket. We also demonstrate that this flexible docking algorithm may be applicable to RNA docking with a representative riboswitch example. Our findings show significant improvements in top ranking accuracy across this set, with the largest improvement relative to rigid, 23.64%, occurring for ligands binding to DHFR. We then evaluate the ability to identify lead compounds among a large chemical space for the proposed flexible receptor docking algorithm using a subset of the DUD-E containing receptor targets MCR, GCR, and ANDR. We demonstrate that our new algorithms show improved performance in modeling flexible binding site residues compared to DOCK. Finally, we select the T4 L99A and T4 L99A/M102Q decoy sets, containing dozens of binders and experimentally validated nonbinders, to test our approach in distinguishing binders from nonbinders. We illustrate that our new algorithms for searching and scoring have superior performance to rigid receptor CDOCKER as well as AutoDock Vina. Finally, we suggest that flexible CDOCKER is sufficiently fast to be utilized in high-throughput docking screens in the context of hierarchical approaches.
Figures
References
-
- Basak SC Chemobioinformatics: the advancing frontier of computer-aided drug design in the post-genomic era. Curr Comput Aided Drug Des 2012, 8, 1–2. - PubMed
-
- Kitchen DB; Decornez H; Furr JR; Bajorath J Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. - PubMed
-
- Yuriev E; Agostino M; Ramsland PA Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 2011, 24, 149–164. - PubMed
-
- Taylor RD; Jewsbury PJ; Essex JW A review of protein-small molecule docking methods. J. Comput. Aided. Mol. Des. 2002, 16, 151–166. - PubMed
-
- Su M; Yang Q; Du Y; Feng G; Liu Z; Li Y; Wang R Comparative assessment of scoring functions: The CASF-2016 update. J. Chem. Inf. Model. 2018, 59, 895–913. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
