

DNA methylation signatures reveal that distinct combinations of transcription factors specify human immune cell epigenetic identity
- PMID: 34706222
- PMCID: PMC9190145
- DOI: 10.1016/j.immuni.2021.10.001
DNA methylation signatures reveal that distinct combinations of transcription factors specify human immune cell epigenetic identity
Erratum in
-
DNA methylation signatures reveal that distinct combinations of transcription factors specify human immune cell epigenetic identity.Immunity. 2022 Jun 14;55(6):1135. doi: 10.1016/j.immuni.2022.05.006. Immunity. 2022. PMID: 35704994 No abstract available.
Abstract
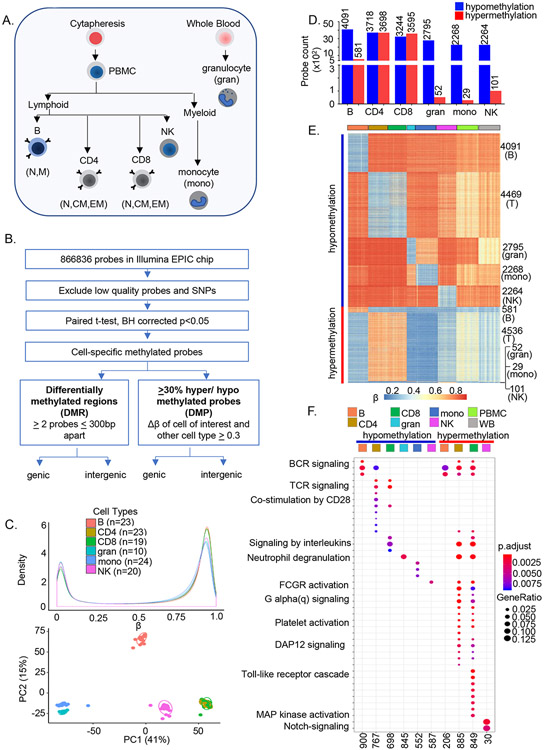
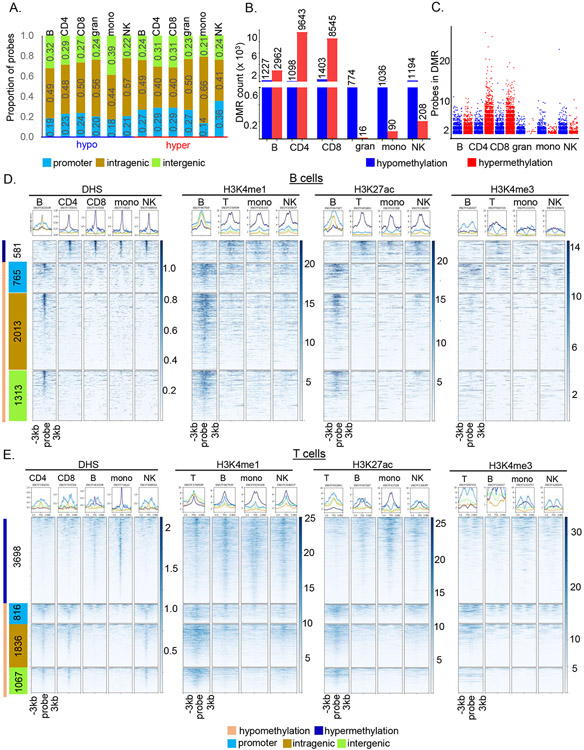
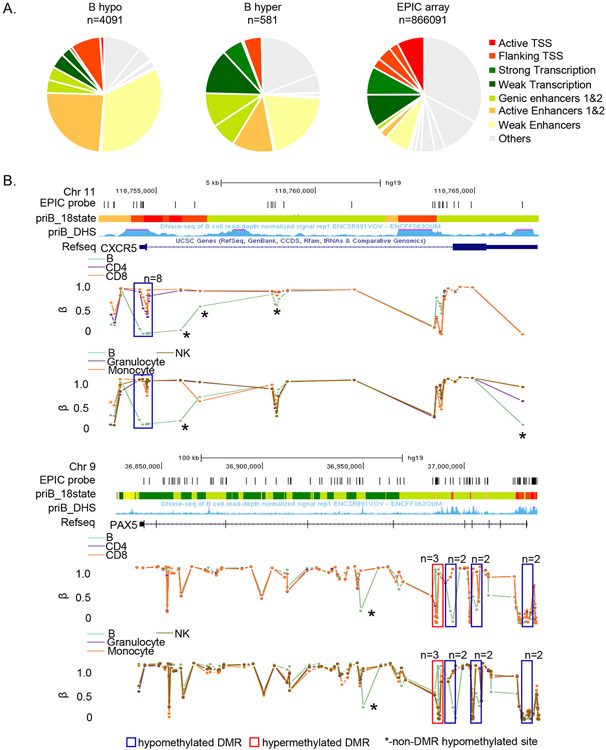
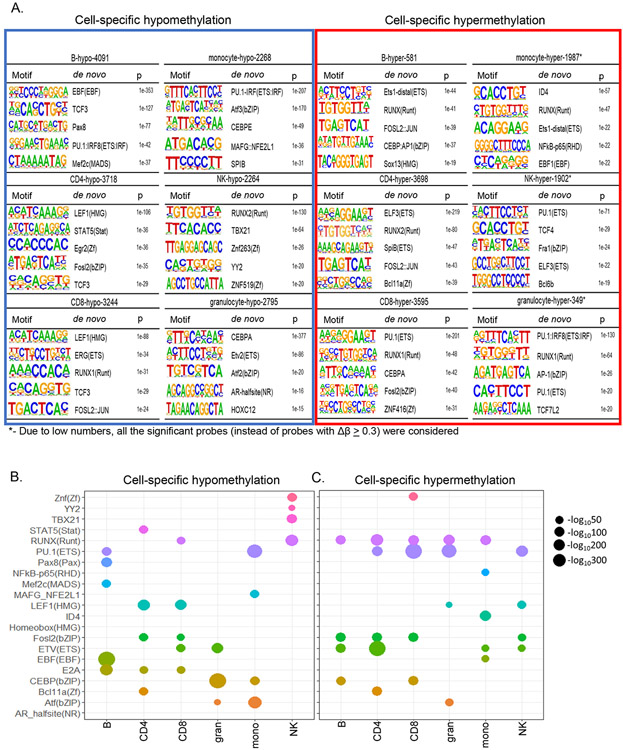
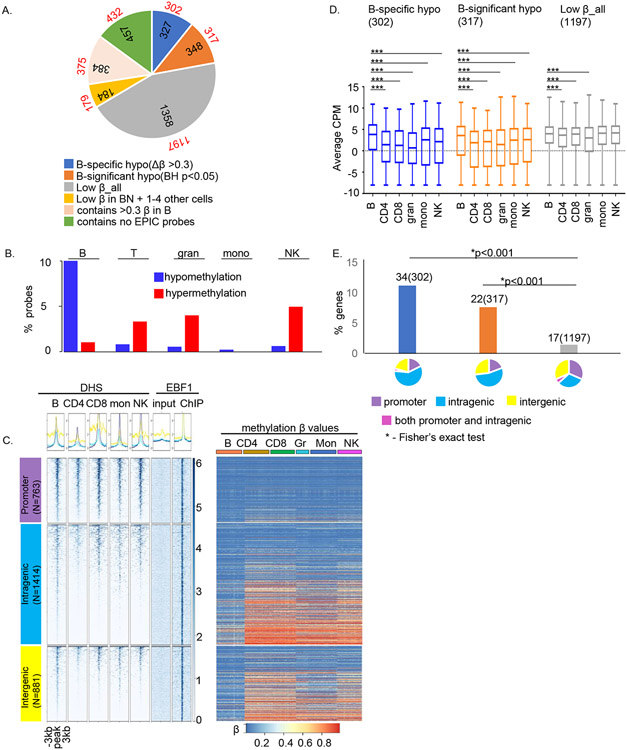
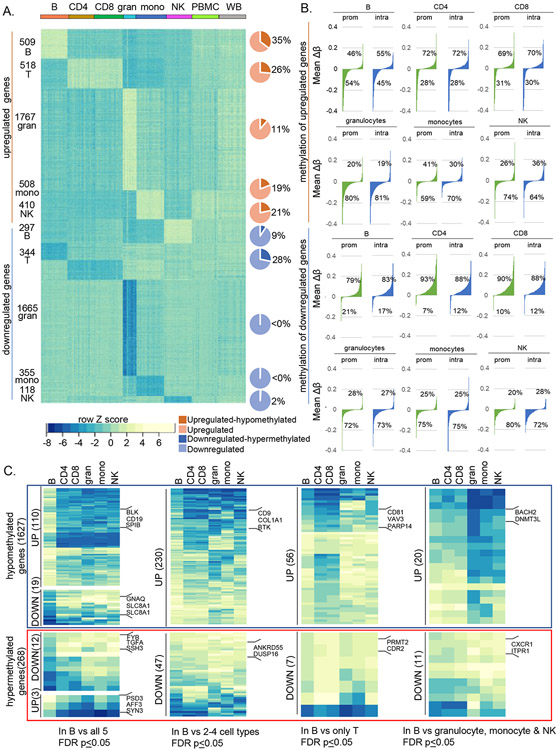
Epigenetic reprogramming underlies specification of immune cell lineages, but patterns that uniquely define immune cell types and the mechanisms by which they are established remain unclear. Here, we identified lineage-specific DNA methylation signatures of six immune cell types from human peripheral blood and determined their relationship to other epigenetic and transcriptomic patterns. Sites of lineage-specific hypomethylation were associated with distinct combinations of transcription factors in each cell type. By contrast, sites of lineage-specific hypermethylation were restricted mostly to adaptive immune cells. PU.1 binding sites were associated with lineage-specific hypo- and hypermethylation in different cell types, suggesting that it regulates DNA methylation in a context-dependent manner. These observations indicate that innate and adaptive immune lineages are specified by distinct epigenetic mechanisms via combinatorial and context-dependent use of key transcription factors. The cell-specific epigenomics and transcriptional patterns identified serve as a foundation for future studies on immune dysregulation in diseases and aging.
Keywords: DNA methylation; epigenetics; gene expression; human immunity; innate cells; lymphocytes; transcription factors.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests A. Battle holds stock in Google and is a consultant for Third Rock Ventures.
Figures
Similar articles
-
Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes.Nucleic Acids Res. 2016 Jan 8;44(1):75-94. doi: 10.1093/nar/gkv1332. Epub 2015 Dec 3. Nucleic Acids Res. 2016. PMID: 26635396 Free PMC article.
-
An integrated epigenomic-transcriptomic landscape of lung cancer reveals novel methylation driver genes of diagnostic and therapeutic relevance.Theranostics. 2021 Mar 11;11(11):5346-5364. doi: 10.7150/thno.58385. eCollection 2021. Theranostics. 2021. PMID: 33859751 Free PMC article.
-
Epigenomics in stress tolerance of plants under the climate change.Mol Biol Rep. 2023 Jul;50(7):6201-6216. doi: 10.1007/s11033-023-08539-6. Epub 2023 Jun 9. Mol Biol Rep. 2023. PMID: 37294468 Review.
-
Distinct Epigenetic Effects of Tobacco Smoking in Whole Blood and among Leukocyte Subtypes.PLoS One. 2016 Dec 9;11(12):e0166486. doi: 10.1371/journal.pone.0166486. eCollection 2016. PLoS One. 2016. PMID: 27935972 Free PMC article.
-
Epigenetics: An opportunity to shape innate and adaptive immune responses.Immunology. 2022 Dec;167(4):451-470. doi: 10.1111/imm.13571. Epub 2022 Sep 26. Immunology. 2022. PMID: 36043705 Review.
Cited by
-
Circulating Histones to Detect and Monitor the Progression of Cancer.Int J Mol Sci. 2023 Jan 4;24(2):942. doi: 10.3390/ijms24020942. Int J Mol Sci. 2023. PMID: 36674455 Free PMC article. Review.
-
Genome-wide DNA methylation-analysis of blastic plasmacytoid dendritic cell neoplasm identifies distinct molecular features.Leukemia. 2024 May;38(5):1086-1098. doi: 10.1038/s41375-024-02240-8. Epub 2024 Apr 10. Leukemia. 2024. PMID: 38600314 Free PMC article.
-
New genetic and epigenetic insights into the chemokine system: the latest discoveries aiding progression toward precision medicine.Cell Mol Immunol. 2023 Jul;20(7):739-776. doi: 10.1038/s41423-023-01032-x. Epub 2023 May 17. Cell Mol Immunol. 2023. PMID: 37198402 Free PMC article. Review.
-
Microarray-Based DNA Methylation Profiling: Validation Considerations for Clinical Testing.J Mol Diagn. 2024 Jun;26(6):447-455. doi: 10.1016/j.jmoldx.2024.02.001. Epub 2024 Feb 18. J Mol Diagn. 2024. PMID: 38378079 Free PMC article.
-
Circulating cell-free DNA methylation patterns indicate cellular sources of allograft injury after liver transplant.Nat Commun. 2025 Jun 17;16(1):5310. doi: 10.1038/s41467-025-60507-9. Nat Commun. 2025. PMID: 40527875 Free PMC article.
References
-
- Andrews S (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data.
-
- Barndt R, Dai MF, and Zhuang Y (1999). A novel role for HEB downstream or parallel to the pre-TCR signaling pathway during alpha beta thymopoiesis. J Immunol 163, 3331–3343. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
