

Crosstalk between autophagy inhibitors and endosome-related secretory pathways: a challenge for autophagy-based treatment of solid cancers
- PMID: 34706732
- PMCID: PMC8549397
- DOI: 10.1186/s12943-021-01423-6
Crosstalk between autophagy inhibitors and endosome-related secretory pathways: a challenge for autophagy-based treatment of solid cancers
Abstract
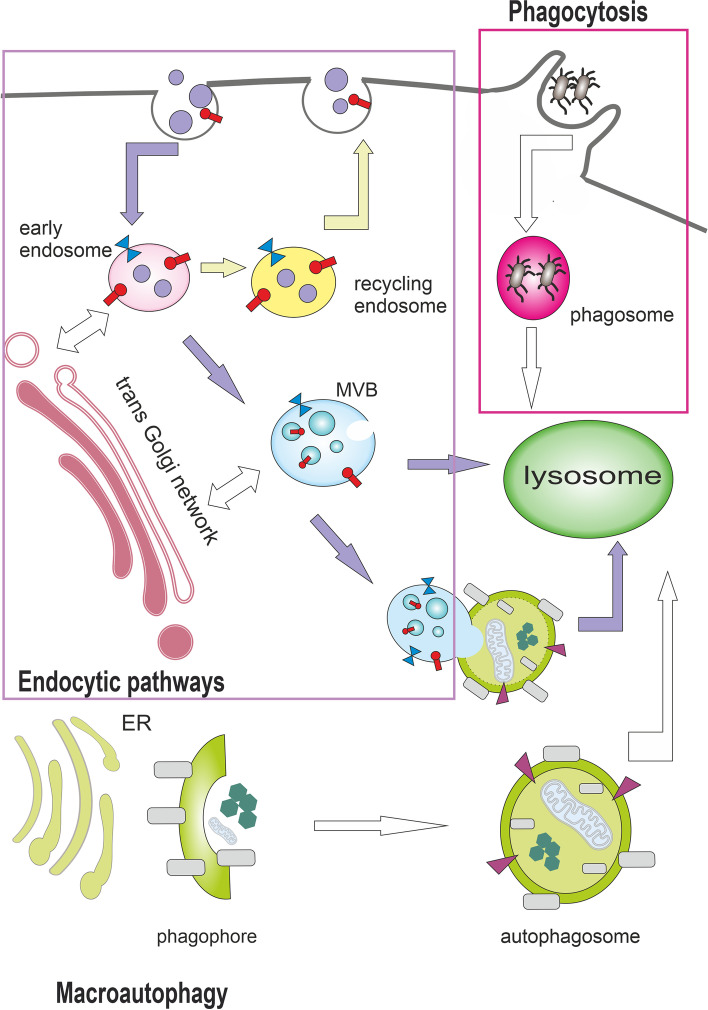
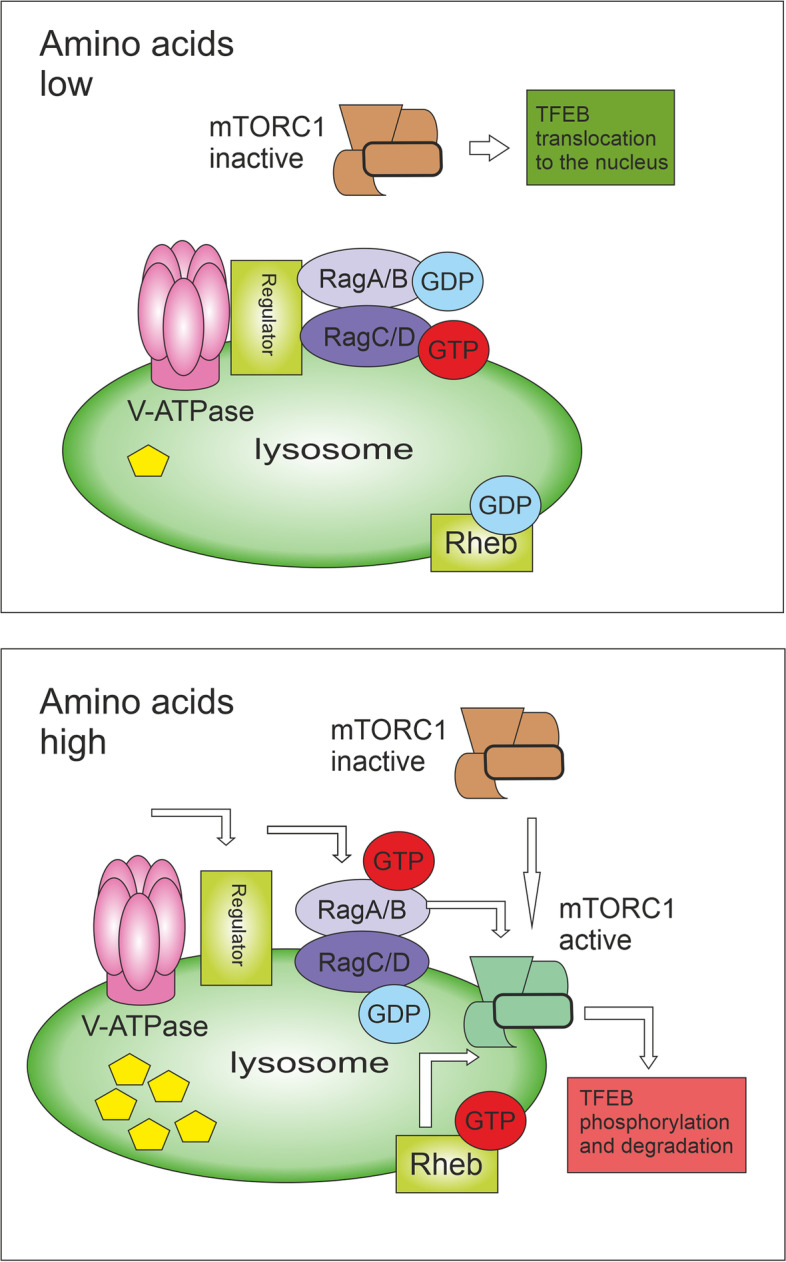
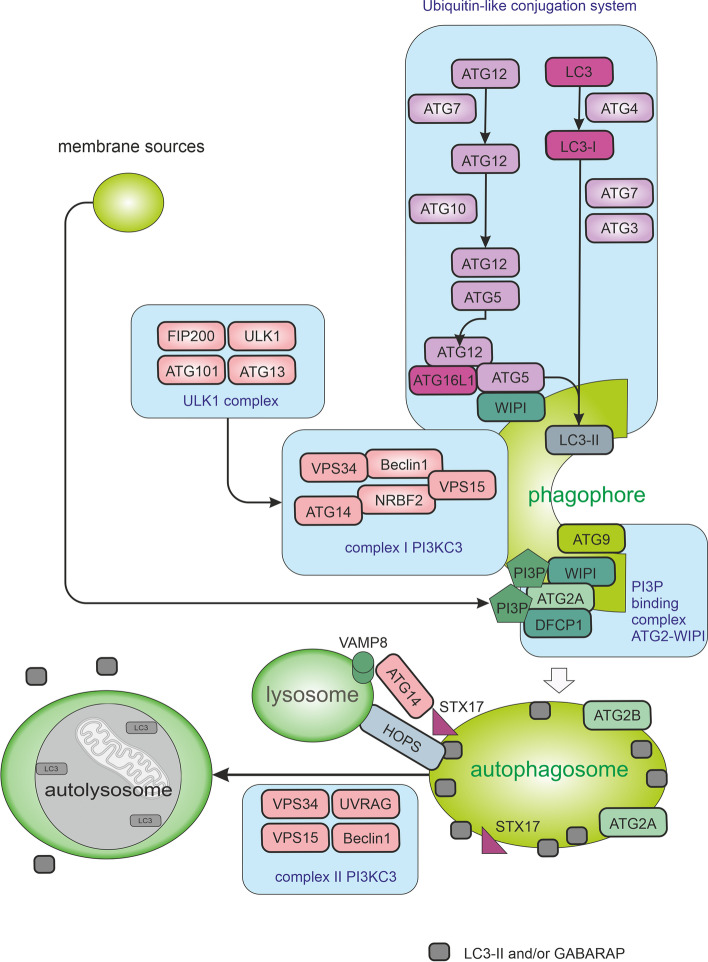
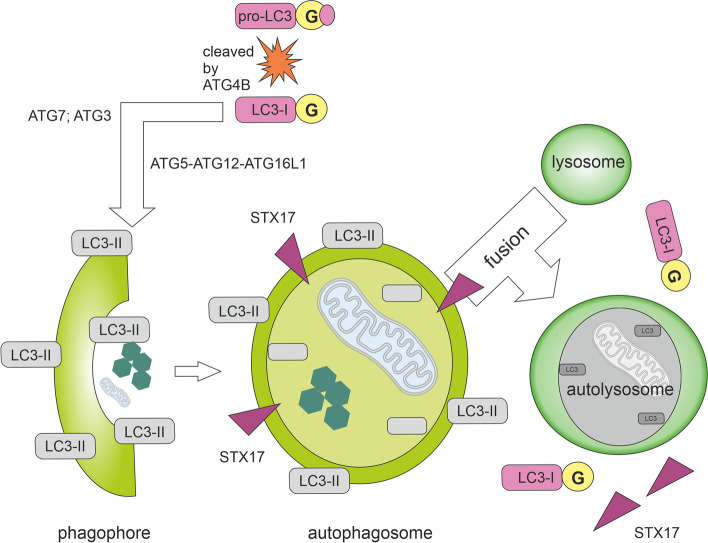
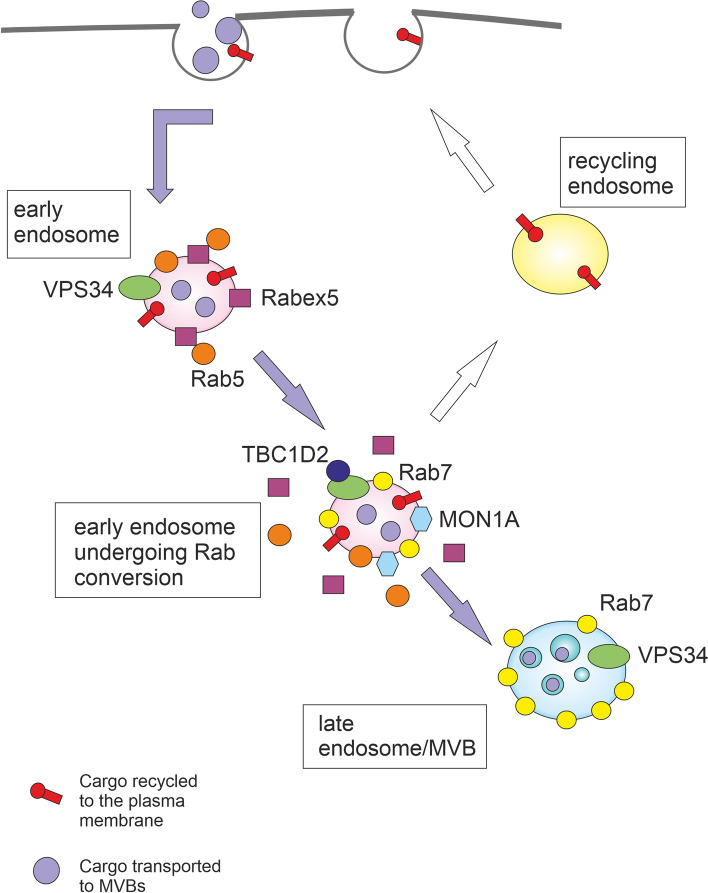
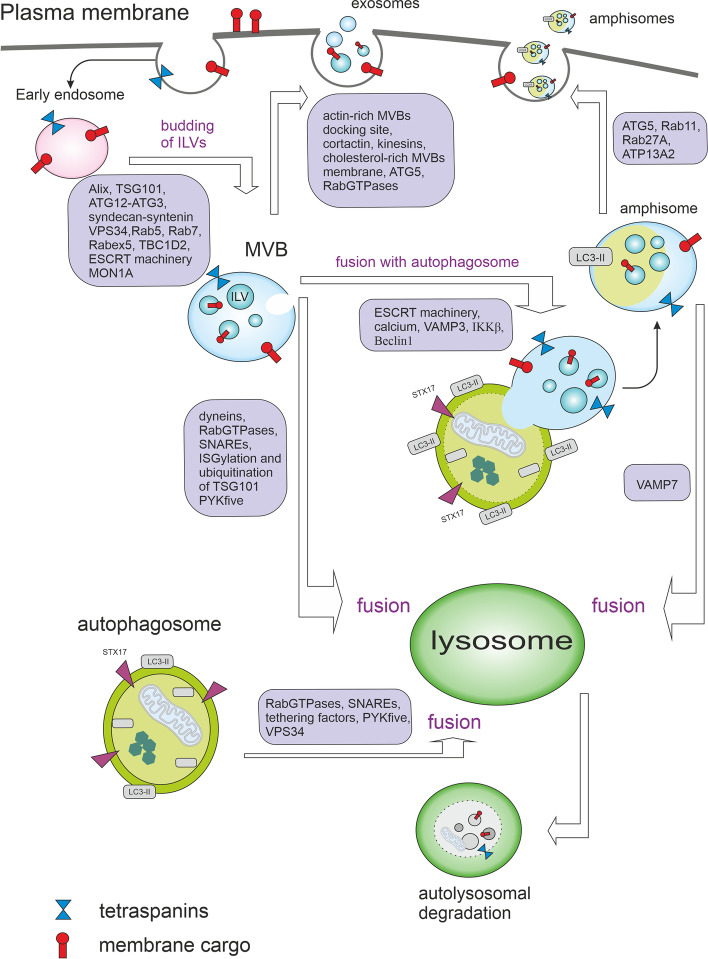
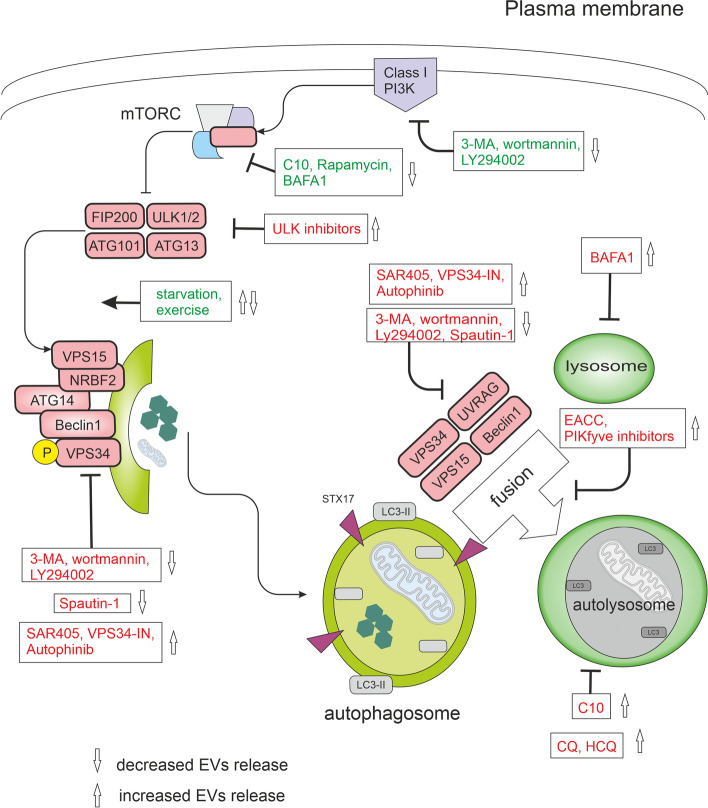
Autophagy is best known for its role in organelle and protein turnover, cell quality control, and metabolism. The autophagic machinery has, however, also adapted to enable protein trafficking and unconventional secretory pathways so that organelles (such as autophagosomes and multivesicular bodies) delivering cargo to lysosomes for degradation can change their mission from fusion with lysosomes to fusion with the plasma membrane, followed by secretion of the cargo from the cell. Some factors with key signalling functions do not enter the conventional secretory pathway but can be secreted in an autophagy-mediated manner.Positive clinical results of some autophagy inhibitors are encouraging. Nevertheless, it is becoming clear that autophagy inhibition, even within the same cancer type, can affect cancer progression differently. Even next-generation inhibitors of autophagy can have significant non-specific effects, such as impacts on endosome-related secretory pathways and secretion of extracellular vesicles (EVs). Many studies suggest that cancer cells release higher amounts of EVs compared to non-malignant cells, which makes the effect of autophagy inhibitors on EVs secretion highly important and attractive for anticancer therapy. In this review article, we discuss how different inhibitors of autophagy may influence the secretion of EVs and summarize the non-specific effects of autophagy inhibitors with a focus on endosome-related secretory pathways. Modulation of autophagy significantly impacts not only the quantity of EVs but also their content, which can have a deep impact on the resulting pro-tumourigenic or anticancer effect of autophagy inhibitors used in the antineoplastic treatment of solid cancers.
Keywords: Amphisomes; Autophagy; Autophagy inhibitors; Cancer; Endosomes; Exosomes; Extracellular vesicles; Multivesicular bodies; Non-conventional secretory pathways.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
