

Chronic viral infections persistently alter marrow stroma and impair hematopoietic stem cell fitness
- PMID: 34709350
- PMCID: PMC8558839
- DOI: 10.1084/jem.20192070
Chronic viral infections persistently alter marrow stroma and impair hematopoietic stem cell fitness
Abstract
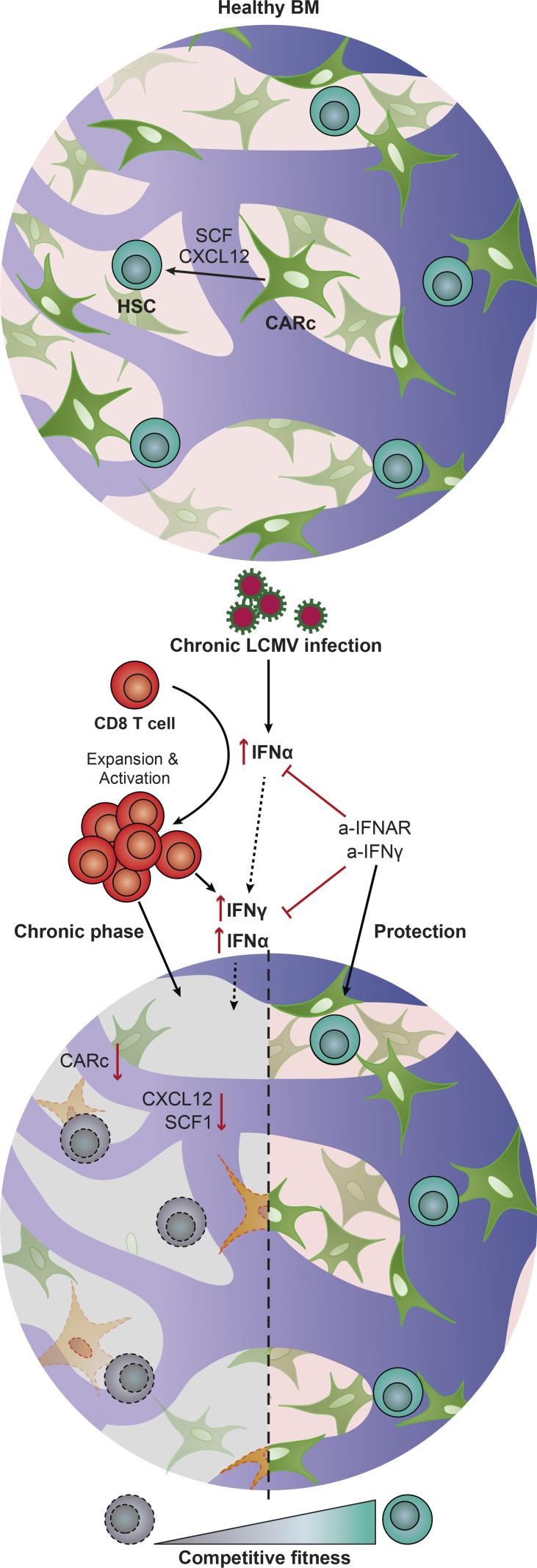
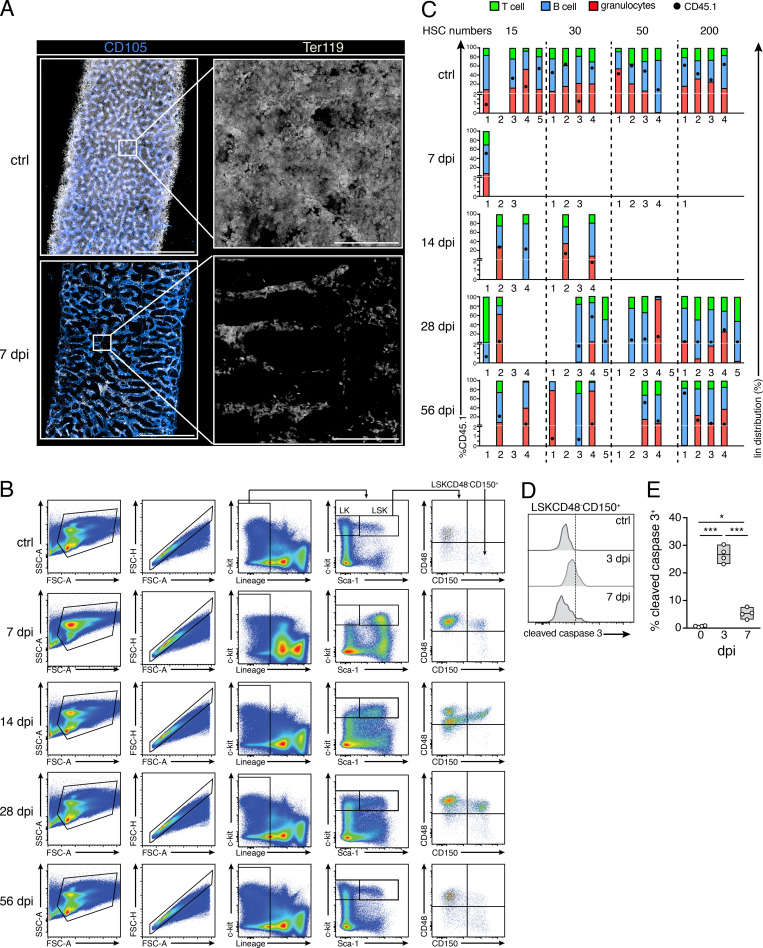
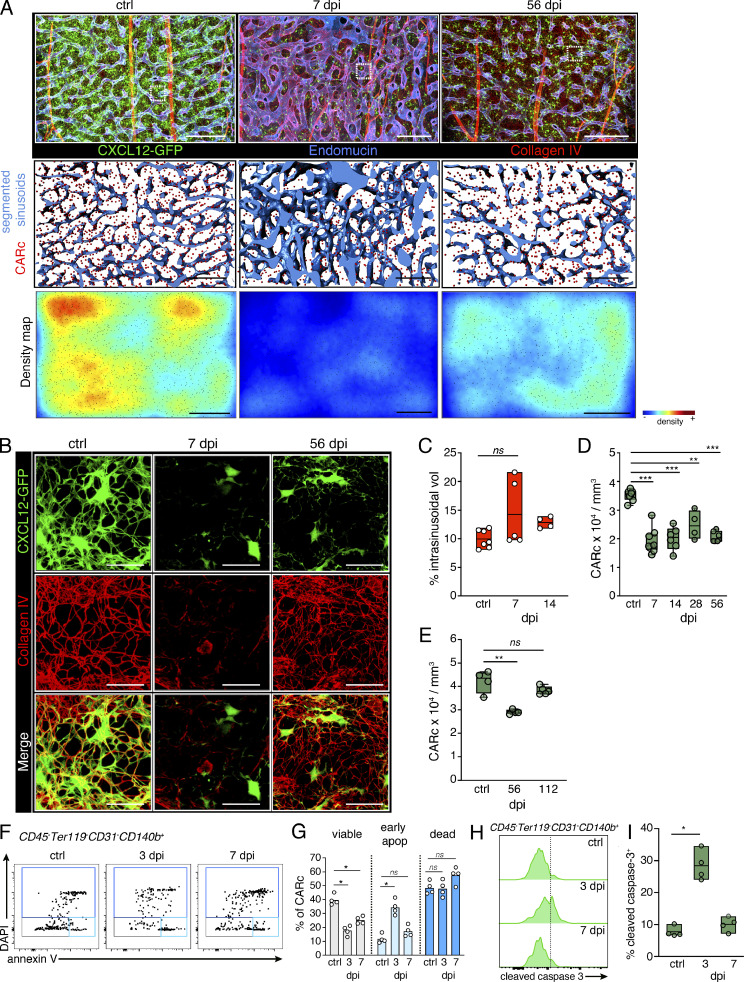
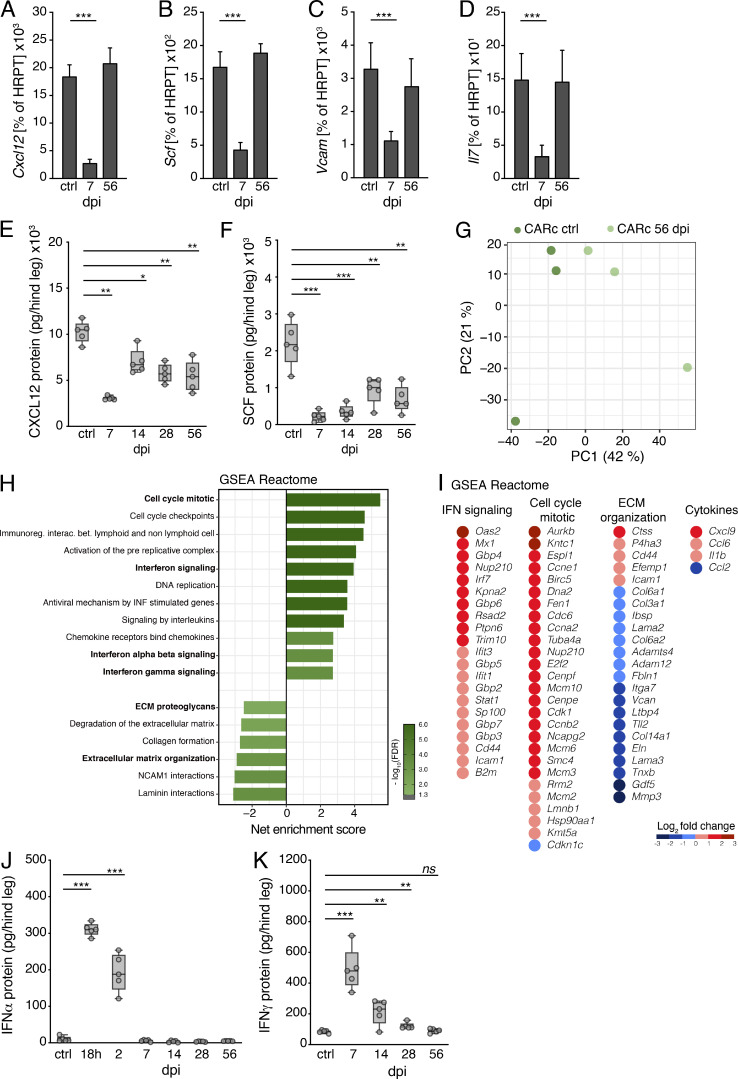
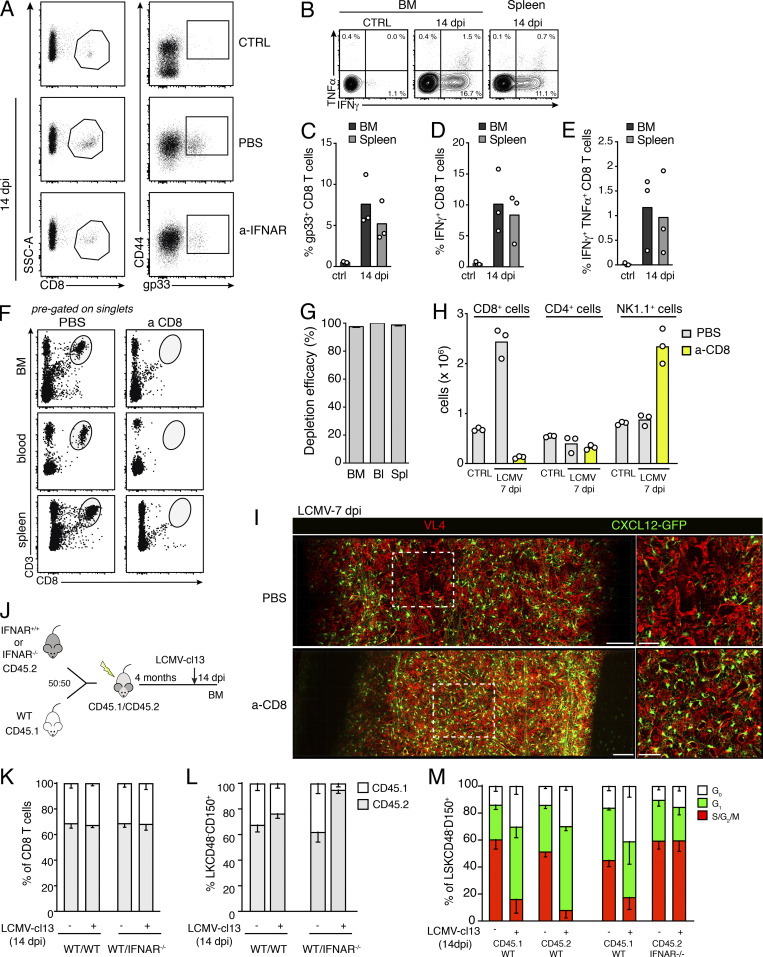
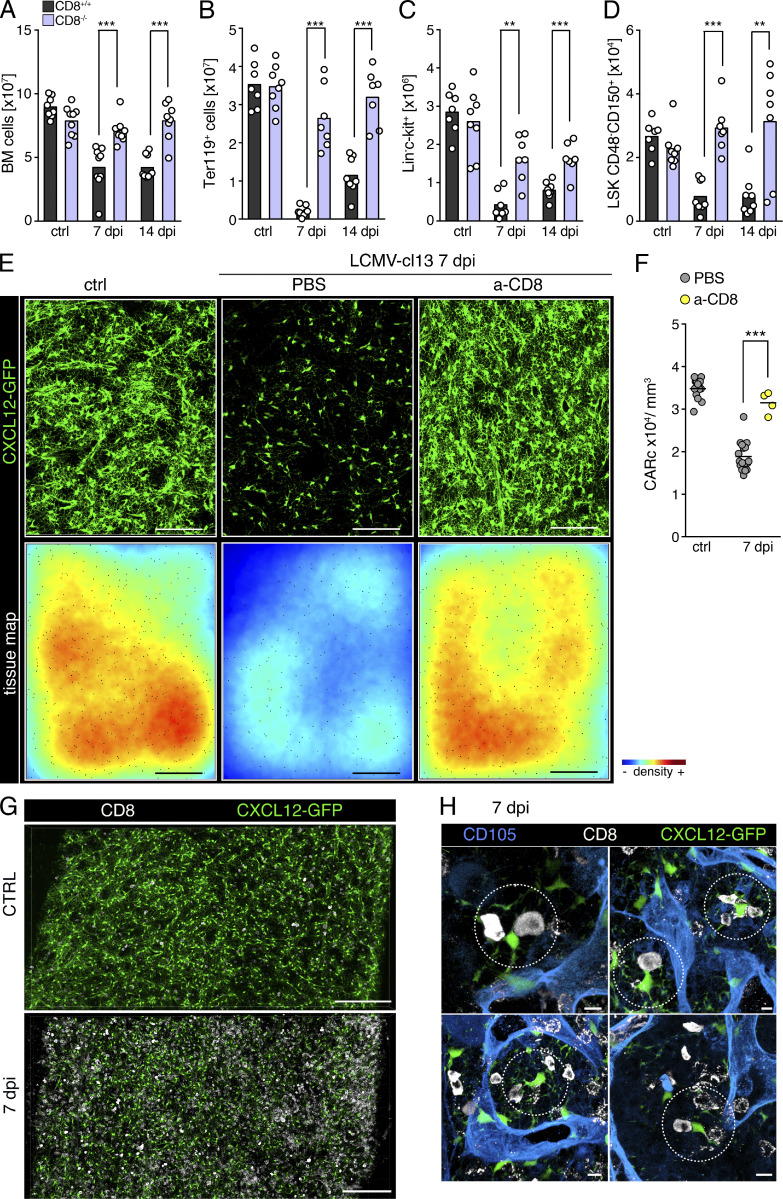
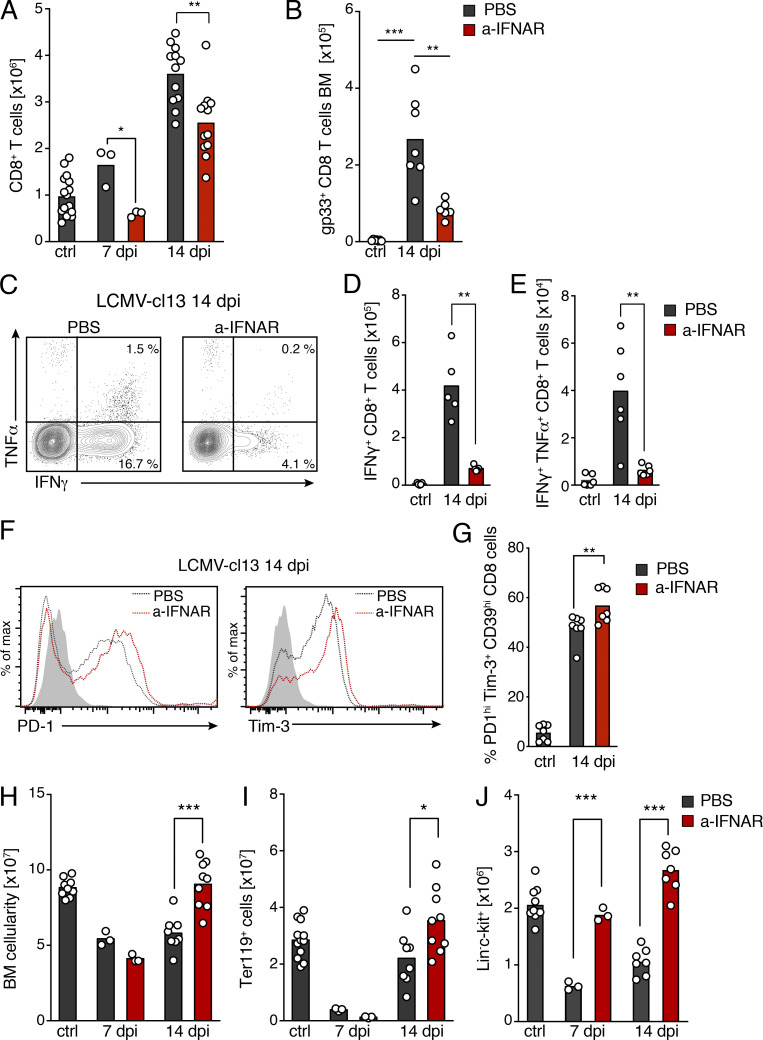
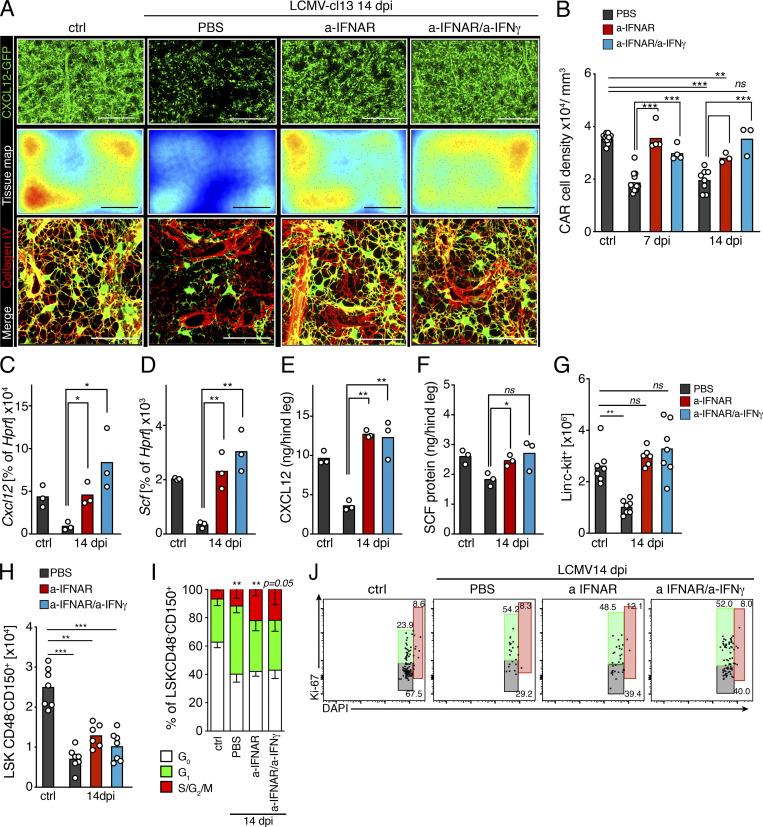
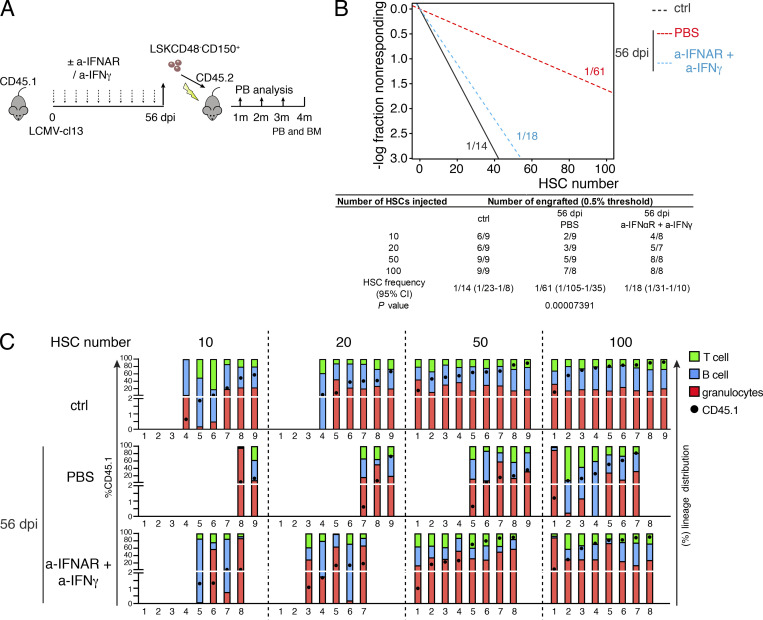
Chronic viral infections are associated with hematopoietic suppression, bone marrow (BM) failure, and hematopoietic stem cell (HSC) exhaustion. However, how persistent viral challenge and inflammatory responses target BM tissues and perturb hematopoietic competence remains poorly understood. Here, we combine functional analyses with advanced 3D microscopy to demonstrate that chronic infection with lymphocytic choriomeningitis virus leads to (1) long-lasting decimation of the BM stromal network of mesenchymal CXCL12-abundant reticular cells, (2) proinflammatory transcriptional remodeling of remaining components of this key niche subset, and (3) durable functional defects and decreased competitive fitness in HSCs. Mechanistically, BM immunopathology is elicited by virus-specific, activated CD8 T cells, which accumulate in the BM via interferon-dependent mechanisms. Combined antibody-mediated inhibition of type I and II IFN pathways completely preempts degeneration of CARc and protects HSCs from chronic dysfunction. Hence, viral infections and ensuing immune reactions durably impact BM homeostasis by persistently decreasing the competitive fitness of HSCs and disrupting essential stromal-derived, hematopoietic-supporting cues.
© 2021 Isringhausen et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures





References
-
- Apperley, J.F., Dowding C., Hibbin J., Buiter J., Matutes E., Sissons P.J., Gordon M., and Goldman J.M.. 1989. The effect of cytomegalovirus on hemopoiesis: in vitro evidence for selective infection of marrow stromal cells. Exp. Hematol. 17:38–45. - PubMed
-
- Baccin, C., Al-Sabah J., Velten L., Helbling P.M., Grünschläger F., Hernández-Malmierca P., Nombela-Arrieta C., Steinmetz L.M., Trumpp A., and Haas S.. 2019. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 22:38–48. 10.1038/s41556-019-0439-6 - DOI - PMC - PubMed
-
- Bahner, I., Kearns K., Coutinho S., Leonard E.H., and Kohn D.B.. 1997. Infection of human marrow stroma by human immunodeficiency virus-1 (HIV-1) is both required and sufficient for HIV-1-induced hematopoietic suppression in vitro: demonstration by gene modification of primary human stroma. Blood. 90:1787–1798. 10.1182/blood.V90.5.1787 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
