

Intracellular Cholesterol Pools Regulate Oncogenic Signaling and Epigenetic Circuitries in Early T-cell Precursor Acute Lymphoblastic Leukemia
- PMID: 34711640
- PMCID: PMC8904296
- DOI: 10.1158/2159-8290.CD-21-0551
Intracellular Cholesterol Pools Regulate Oncogenic Signaling and Epigenetic Circuitries in Early T-cell Precursor Acute Lymphoblastic Leukemia
Abstract
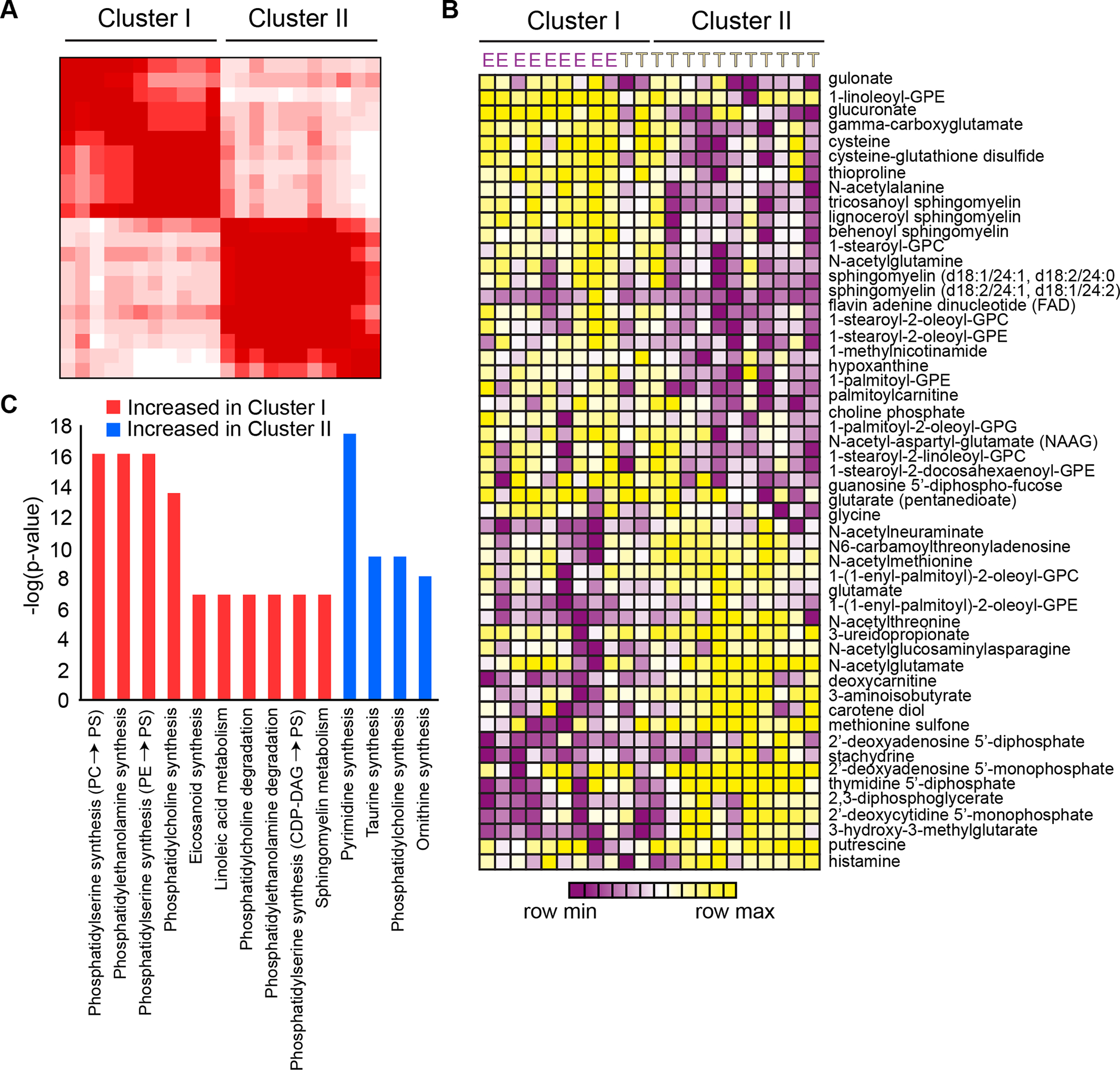
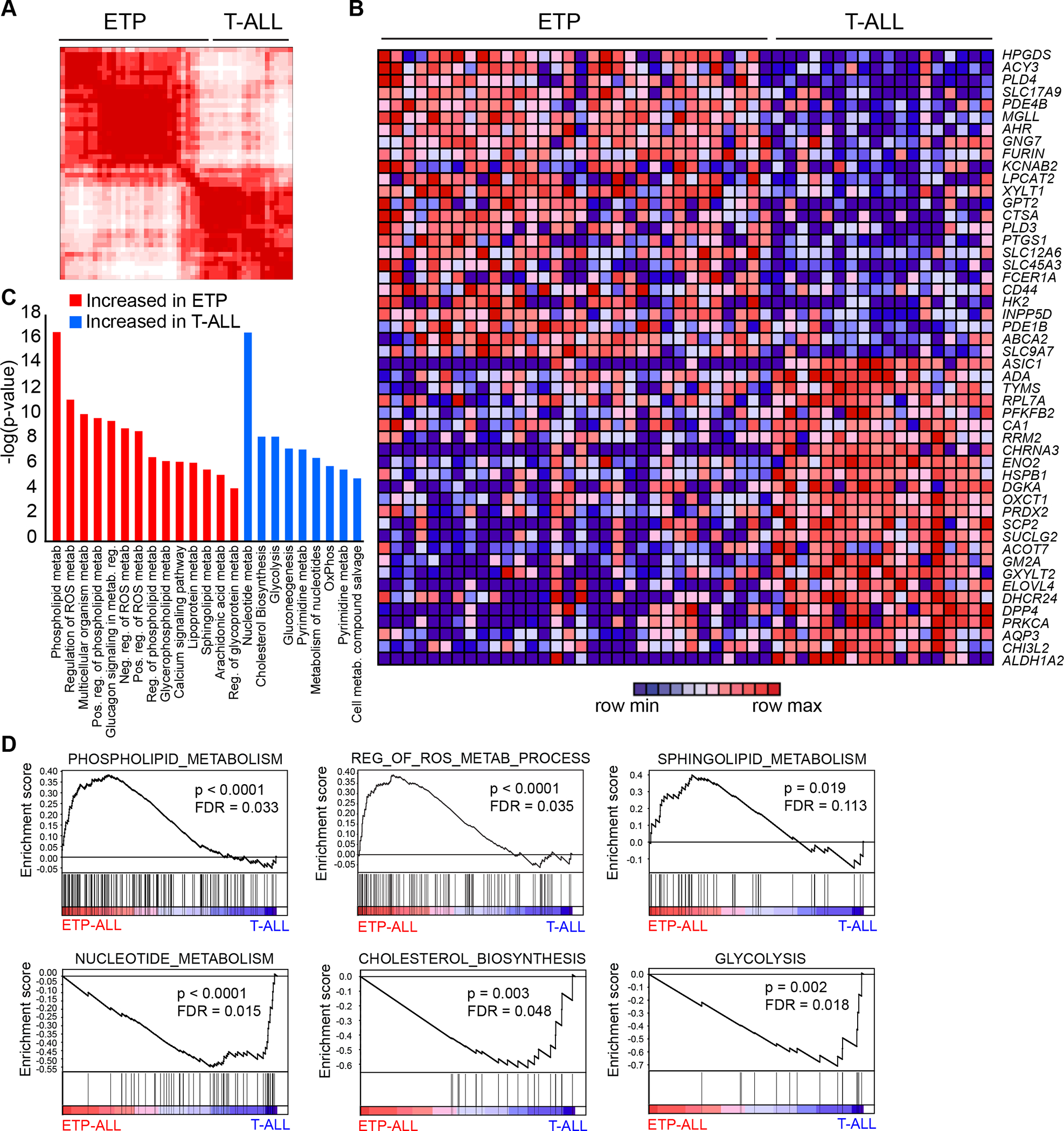
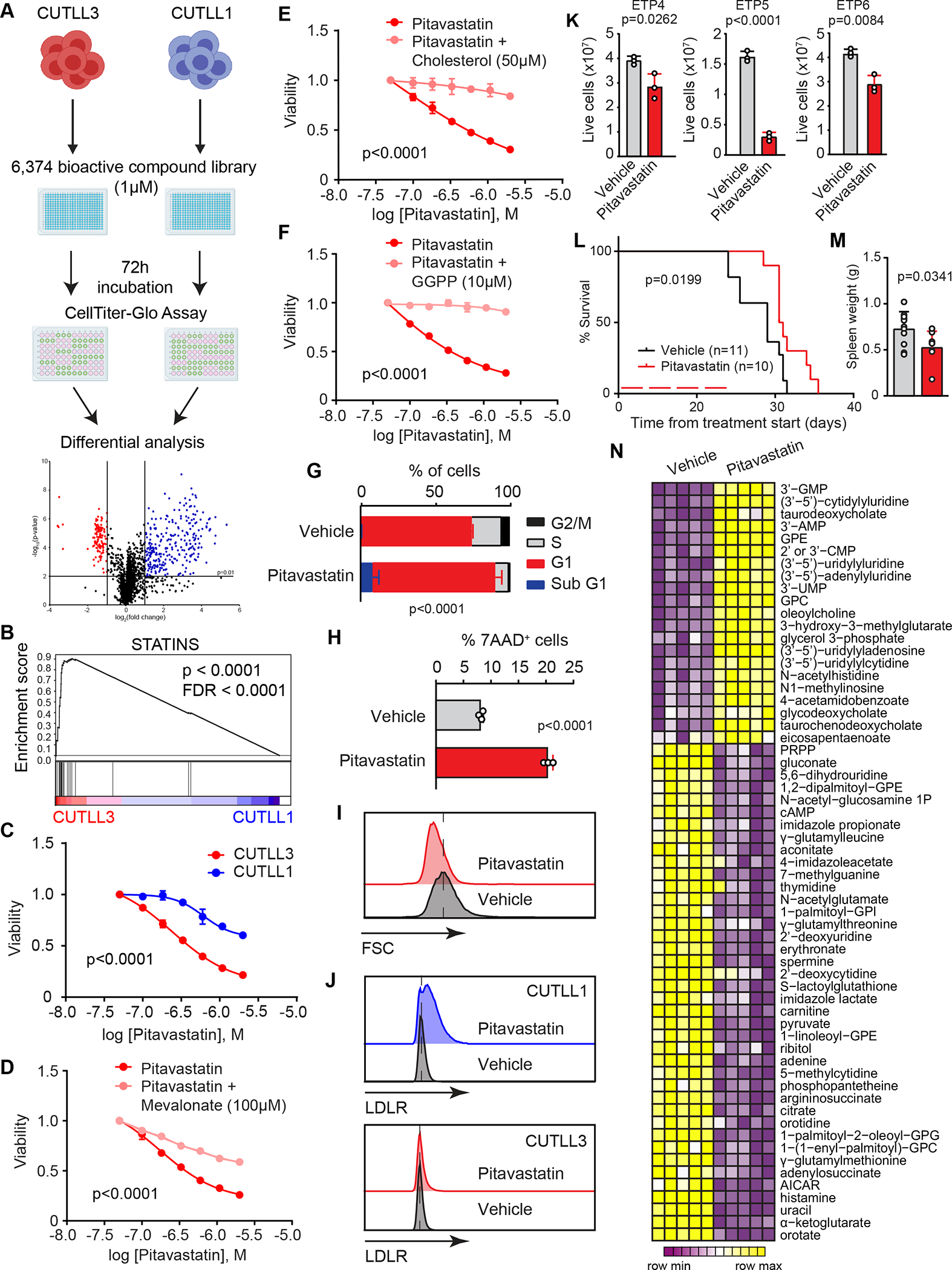
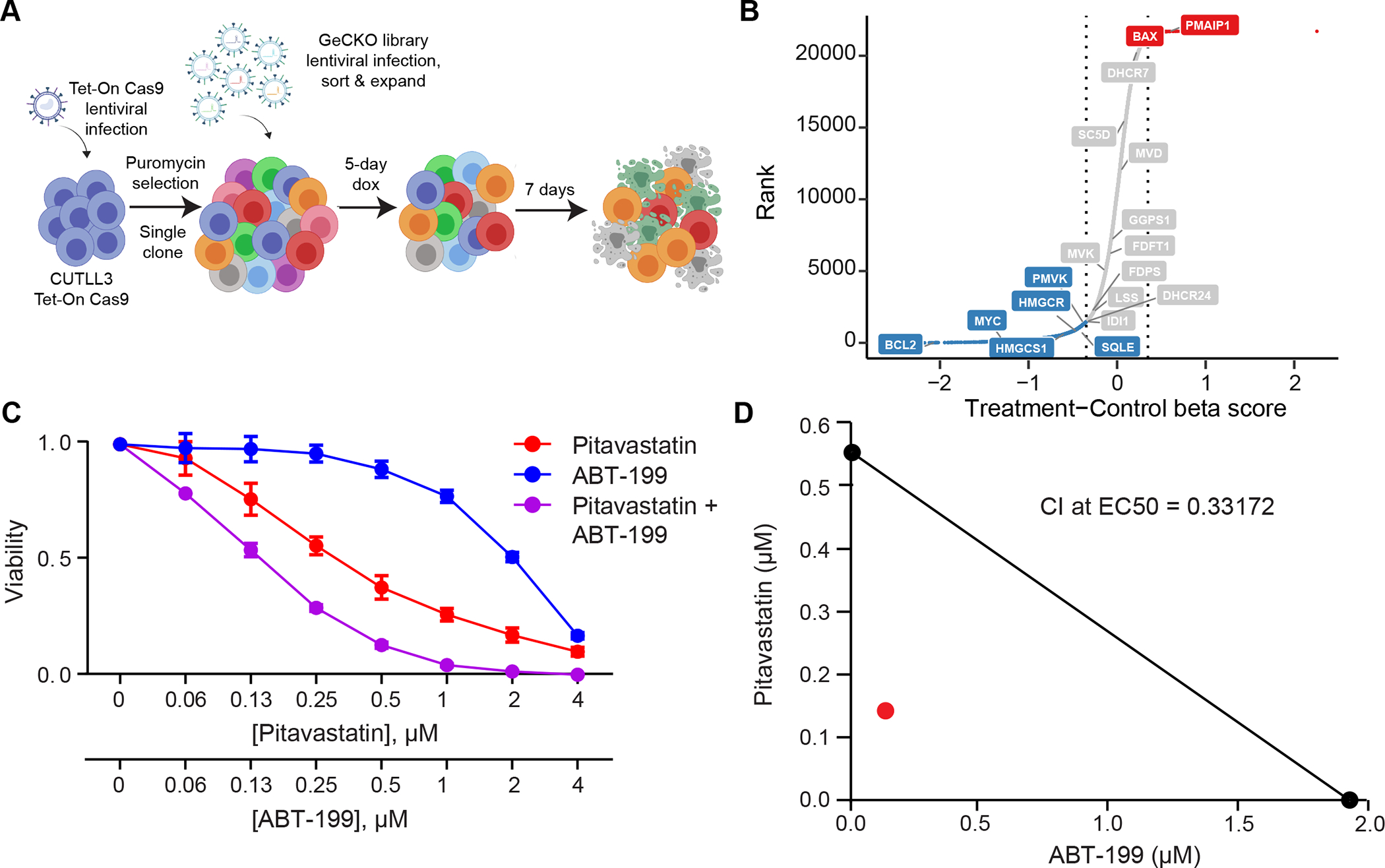
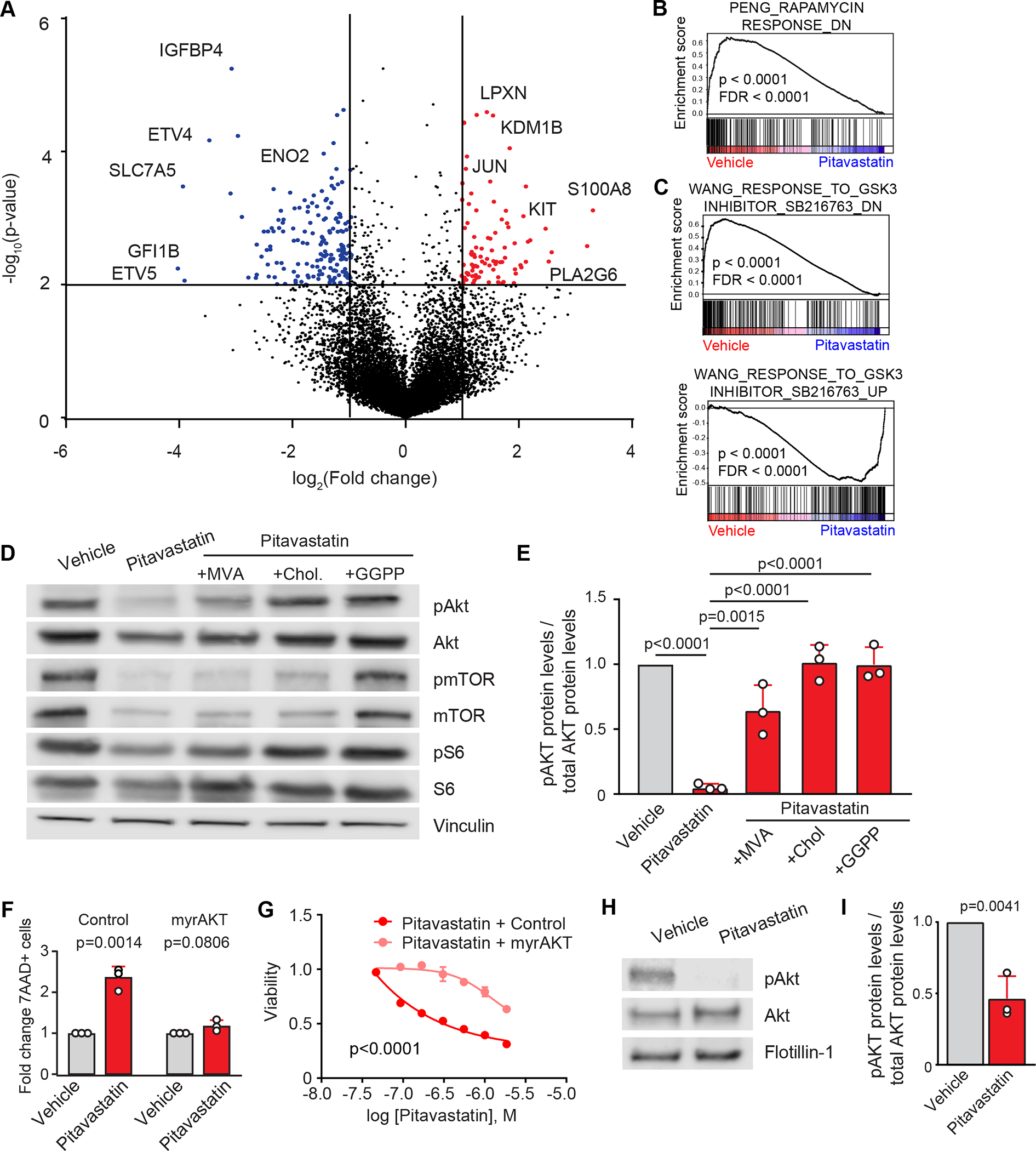
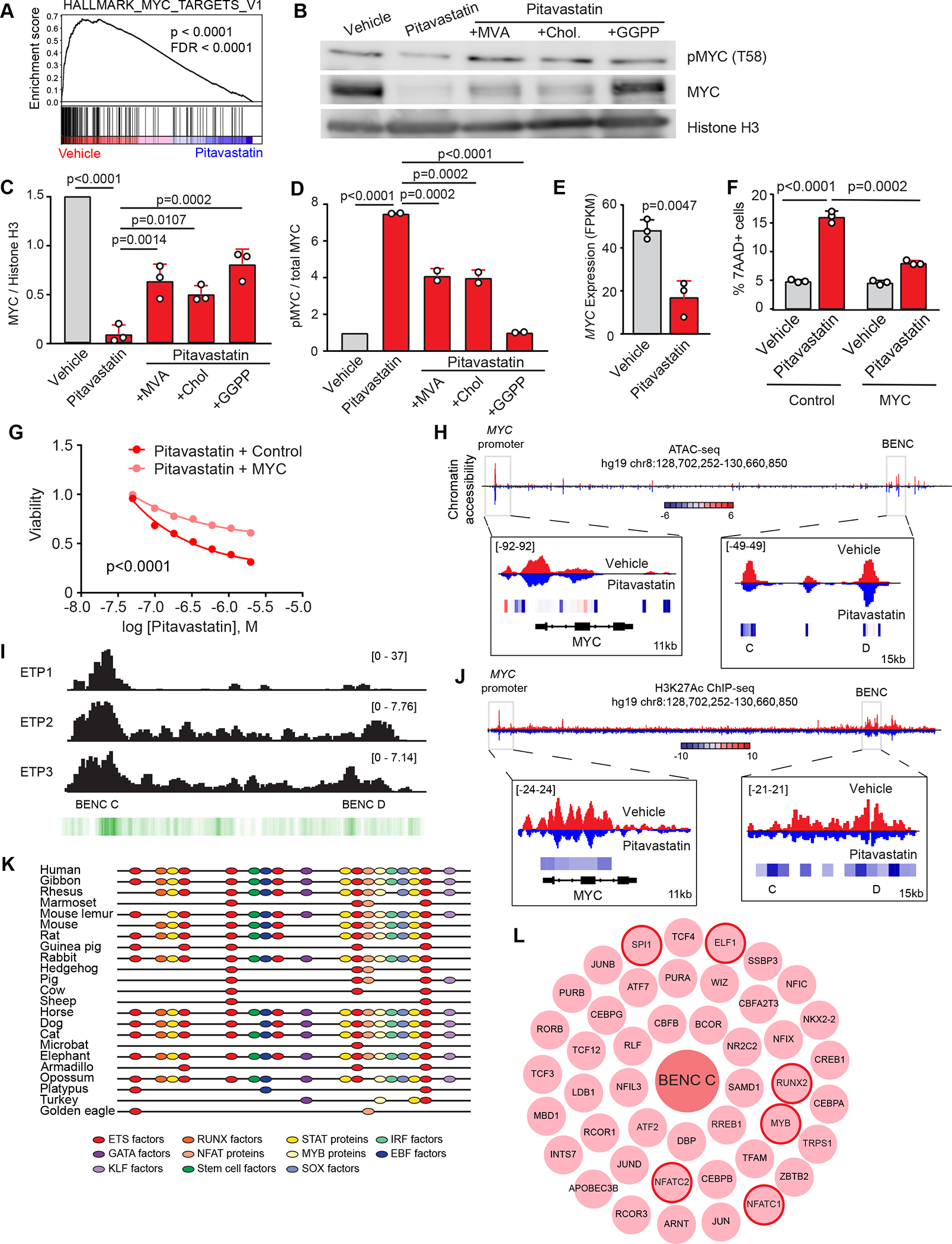
Early T-cell acute lymphoblastic leukemia (ETP-ALL) is an aggressive hematologic malignancy associated with early relapse and poor prognosis that is genetically, immunophenotypically, and transcriptionally distinct from more mature T-cell acute lymphoblastic leukemia (T-ALL) tumors. Here, we leveraged global metabolomic and transcriptomic profiling of primary ETP- and T-ALL leukemia samples to identify specific metabolic circuitries differentially active in this high-risk leukemia group. ETP-ALLs showed increased biosynthesis of phospholipids and sphingolipids and were specifically sensitive to inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase, the rate-limiting enzyme in the mevalonate pathway. Mechanistically, inhibition of cholesterol synthesis inhibited oncogenic AKT1 signaling and suppressed MYC expression via loss of chromatin accessibility at a leukemia stem cell-specific long-range MYC enhancer. In all, these results identify the mevalonate pathway as a druggable novel vulnerability in high-risk ETP-ALL cells and uncover an unanticipated critical role for cholesterol biosynthesis in signal transduction and epigenetic circuitries driving leukemia cell growth and survival.
Significance: Overtly distinct cell metabolic pathways operate in ETP- and T-ALL pointing to specific metabolic vulnerabilities. Inhibition of mevalonate biosynthesis selectively blocks oncogenic AKT-MYC signaling in ETP-ALL and suppresses leukemia cell growth. Ultimately, these results will inform the development of novel tailored and more effective treatments for patients with high-risk ETP-ALL. This article is highlighted in the In This Issue feature, p. 587.
©2021 American Association for Cancer Research.
Conflict of interest statement
Declaration of Interests
The authors declare no competing financial interests relevant for the work reported here. Financial disclosures for Adolfo Ferrando: Consulting for Ayala Pharmaceuticals and SpringWorks Therapeutics; previous research support by Pfizer, Bristol Myers Squib, Merck, Eli Lilly; patent and reagent licensing royalties from Novartis, EMD Millipore and Applied Biological Materials.
Figures
References
-
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proceedings of the National Academy of Sciences of the United States of America 2006;103(48):18261–6 doi 10.1073/pnas.0606108103. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
