

An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci
- PMID: 34711957
- PMCID: PMC7611956
- DOI: 10.1038/s41588-021-00945-5
An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci
Abstract
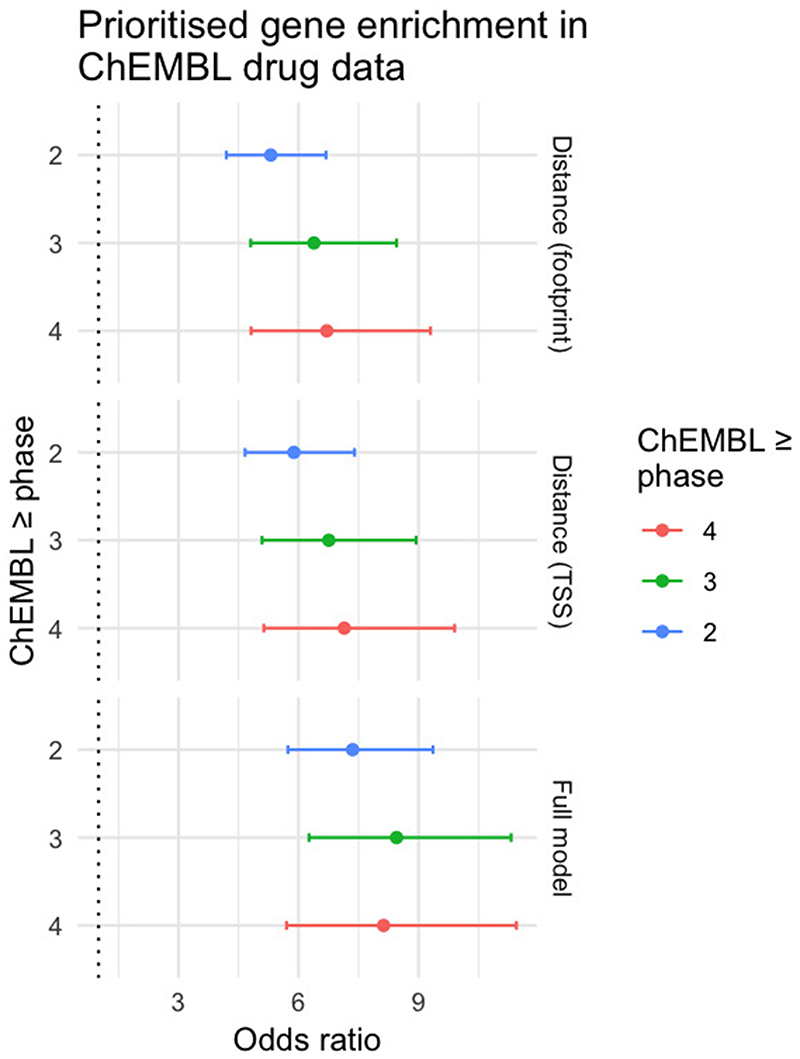
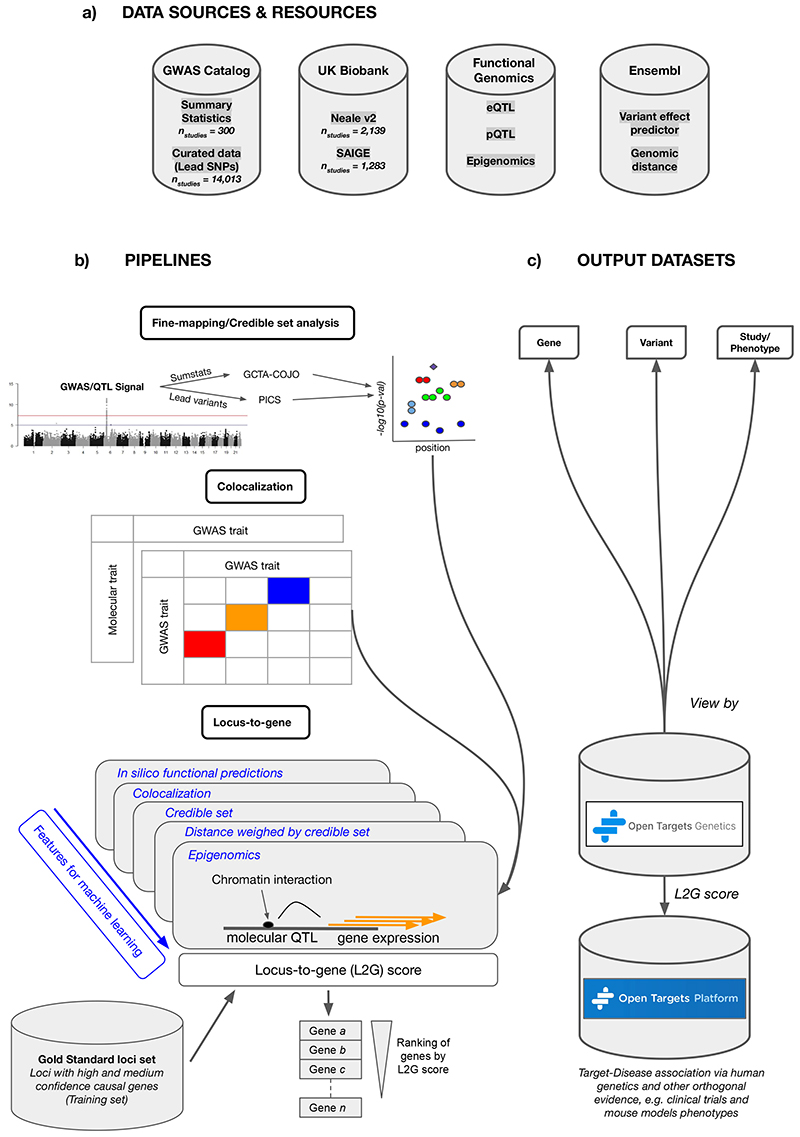
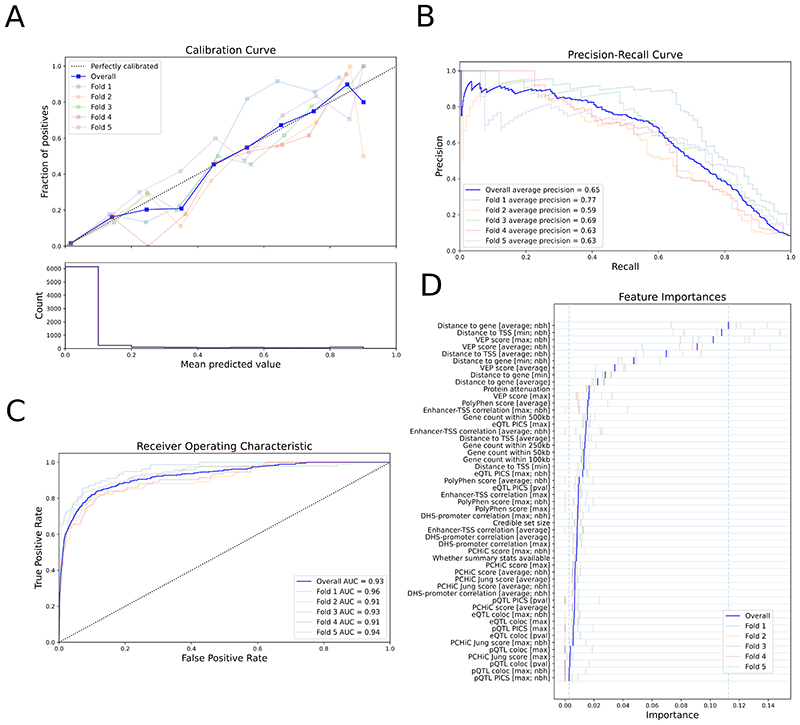
Genome-wide association studies (GWASs) have identified many variants associated with complex traits, but identifying the causal gene(s) is a major challenge. In the present study, we present an open resource that provides systematic fine mapping and gene prioritization across 133,441 published human GWAS loci. We integrate genetics (GWAS Catalog and UK Biobank) with transcriptomic, proteomic and epigenomic data, including systematic disease-disease and disease-molecular trait colocalization results across 92 cell types and tissues. We identify 729 loci fine mapped to a single-coding causal variant and colocalized with a single gene. We trained a machine-learning model using the fine-mapped genetics and functional genomics data and 445 gold-standard curated GWAS loci to distinguish causal genes from neighboring genes, outperforming a naive distance-based model. Our prioritized genes were enriched for known approved drug targets (odds ratio = 8.1, 95% confidence interval = 5.7, 11.5). These results are publicly available through a web portal ( http://genetics.opentargets.org ), enabling users to easily prioritize genes at disease-associated loci and assess their potential as drug targets.
© 2021. Crown.
Conflict of interest statement
J.A.T. is a member of the GSK Human Genetics Advisory Board. E.B.F. is a full time employee of and shareholder in Pfizer, Inc. E.P. was an employee of Biogen at the time of the work. E.P. is now an employee of AstraZeneca.
Figures
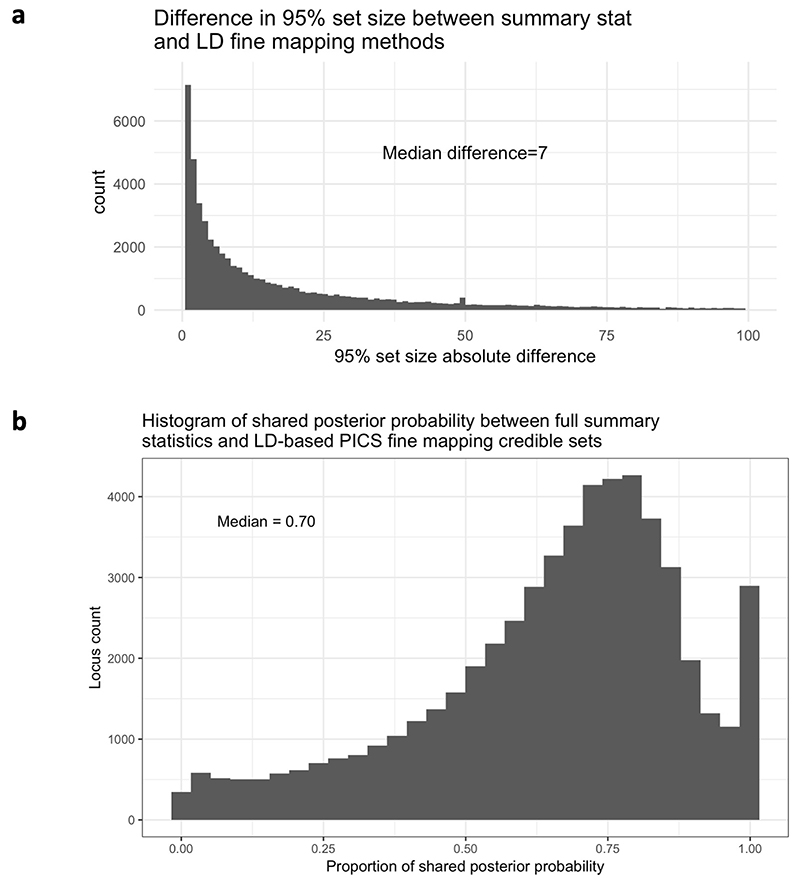
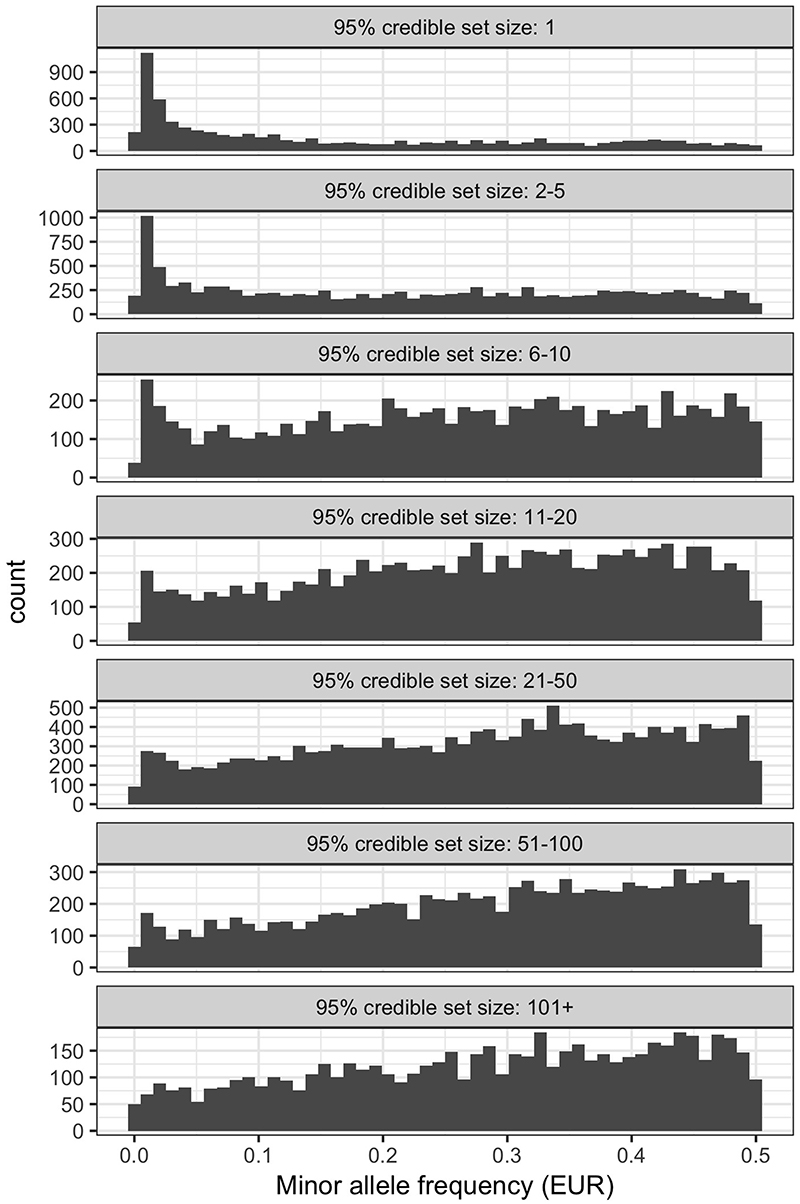
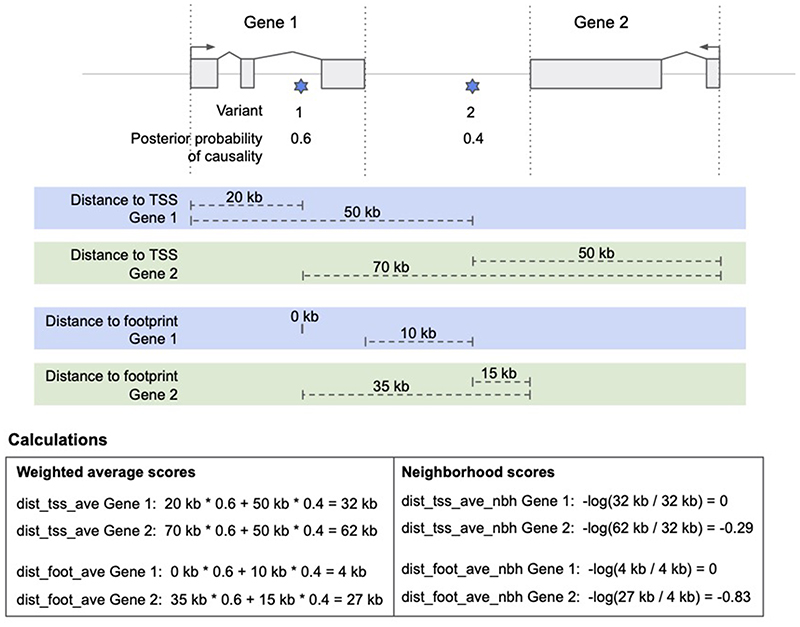
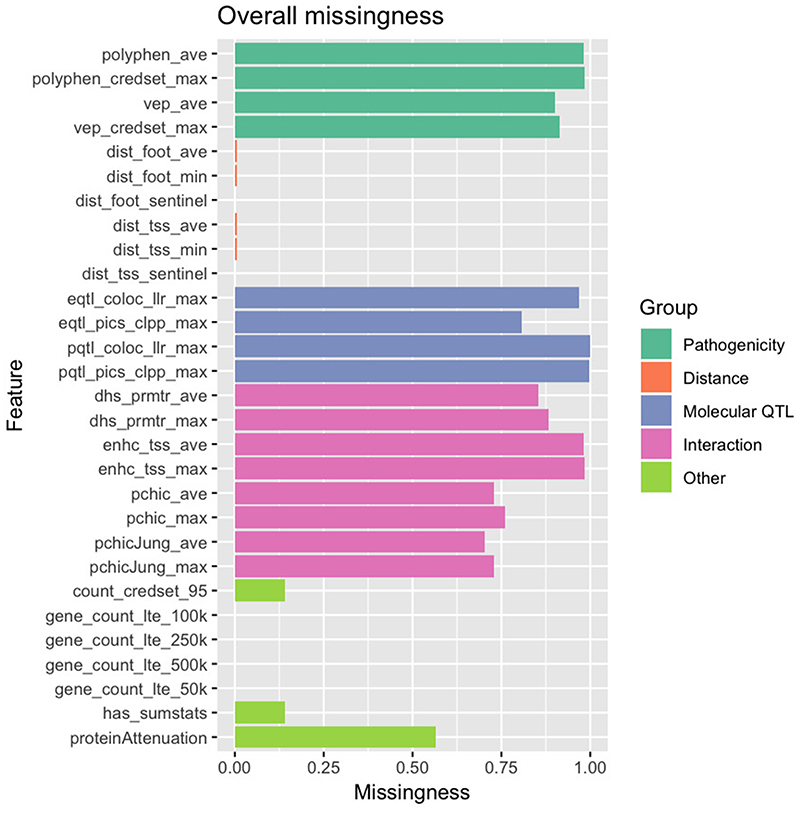
References
-
- Zhu Z, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–487. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
