

Proximity Extension Assay in Combination with Next-Generation Sequencing for High-throughput Proteome-wide Analysis
- PMID: 34715355
- PMCID: PMC8633680
- DOI: 10.1016/j.mcpro.2021.100168
Proximity Extension Assay in Combination with Next-Generation Sequencing for High-throughput Proteome-wide Analysis
Abstract
Understanding the dynamics of the human proteome is crucial for developing biomarkers to be used as measurable indicators for disease severity and progression, patient stratification, and drug development. The Proximity Extension Assay (PEA) is a technology that translates protein information into actionable knowledge by linking protein-specific antibodies to DNA-encoded tags. In this report we demonstrate how we have combined the unique PEA technology with an innovative and automated sample preparation and high-throughput sequencing readout enabling parallel measurement of nearly 1500 proteins in 96 samples generating close to 150,000 data points per run. This advancement will have a major impact on the discovery of new biomarkers for disease prediction and prognosis and contribute to the development of the rapidly evolving fields of wellness monitoring and precision medicine.
Keywords: antibody; biomarker; immunoassay; multiplex; next-generation sequencing; plasma; proteomics; proximity extension assay; serum.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest All authors are employees of Olink Proteomics AB commercializing the described method. An author may or may not be named as an inventor on current patents owned and controlled by Olink Proteomics AB, https://www.olink.com/patents/, as amended from time to time. All authors have financial interest in Olink Holding AB.
Figures






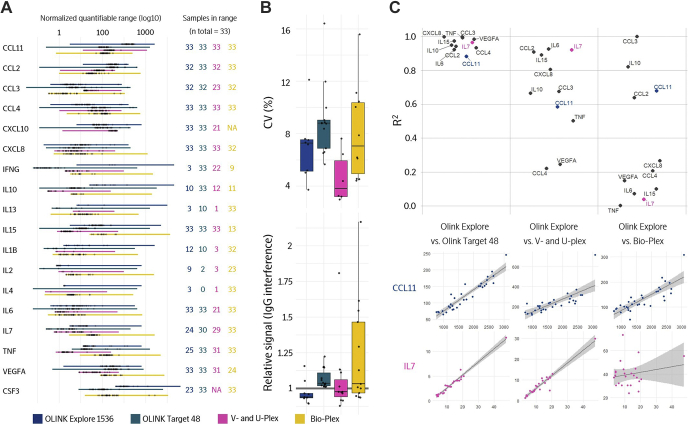


References
-
- Hortin G.L., Sviridov D. The dynamic range problem in the analysis of the plasma proteome. J. Proteomics. 2010;73:629–636. - PubMed
-
- Anderson N.L., Anderson N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteomics. 2002;1:845–867. - PubMed
-
- Joshi A., Rienks M., Theofilatos K., Mayr M. Systems biology in cardiovascular disease: A multiomics approach. Nat. Rev. Cardiol. 2021;18:313–330. - PubMed
-
- Assarsson E., Lundberg M., Holmquist G., Björkesten J., Thorsen S.B., Ekman D., Eriksson A., Rennel Dickens E., Ohlsson S., Edfeldt G., Andersson A.C., Lindstedt P., Stenvang J., Gullberg M., Fredriksson S. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9(4) - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
