

Detection of PKD1 and PKD2 Somatic Variants in Autosomal Dominant Polycystic Kidney Cyst Epithelial Cells by Whole-Genome Sequencing
- PMID: 34716216
- PMCID: PMC8638386
- DOI: 10.1681/ASN.2021050690
Detection of PKD1 and PKD2 Somatic Variants in Autosomal Dominant Polycystic Kidney Cyst Epithelial Cells by Whole-Genome Sequencing
Abstract
Background: Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disorder characterized by the development of multiple cysts in the kidneys. It is often caused by pathogenic mutations in PKD1 and PKD2 genes that encode polycystin proteins. Although the molecular mechanisms for cystogenesis are not established, concurrent inactivating germline and somatic mutations in PKD1 and PKD2 have been previously observed in renal tubular epithelium (RTE).
Methods: To further investigate the cellular recessive mechanism of cystogenesis in RTE, we conducted whole-genome DNA sequencing analysis to identify germline variants and somatic alterations in RTE of 90 unique kidney cysts obtained during nephrectomy from 24 unrelated participants.
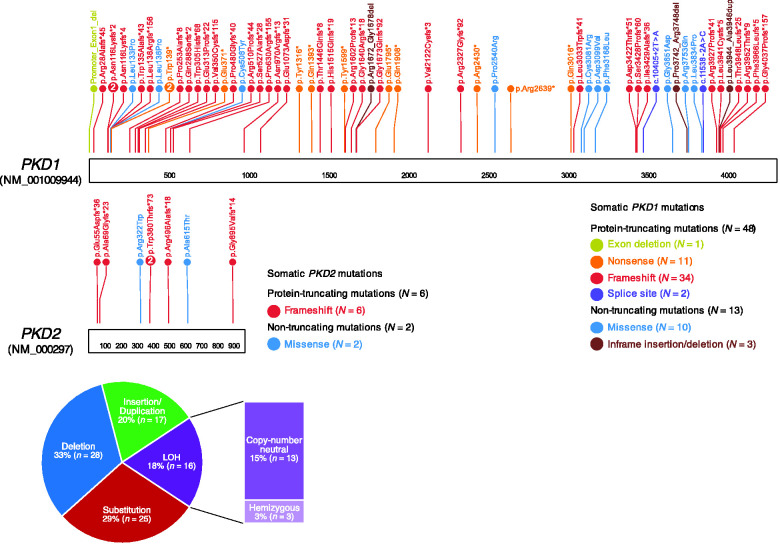
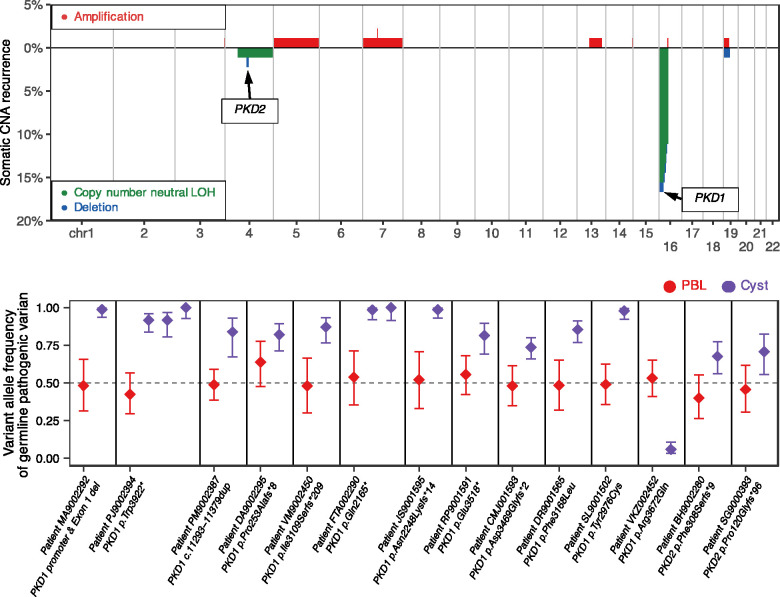
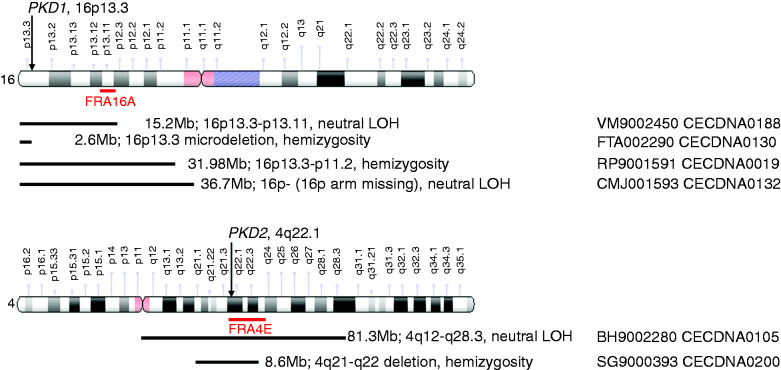
Results: Kidney cysts were overall genomically stable, with low burdens of somatic short mutations or large-scale structural alterations. Pathogenic somatic "second hit" alterations disrupting PKD1 or PKD2 were identified in 93% of the cysts. Of these, 77% of cysts acquired short mutations in PKD1 or PKD2 ; specifically, 60% resulted in protein truncations (nonsense, frameshift, or splice site) and 17% caused non-truncating mutations (missense, in-frame insertions, or deletions). Another 18% of cysts acquired somatic chromosomal loss of heterozygosity (LOH) events encompassing PKD1 or PKD2 ranging from 2.6 to 81.3 Mb. 14% of these cysts harbored copy number neutral LOH events, while the other 3% had hemizygous chromosomal deletions. LOH events frequently occurred at chromosomal fragile sites, or in regions comprising chromosome microdeletion diseases/syndromes. Almost all somatic "second hit" alterations occurred at the same germline mutated PKD1/2 gene.
Conclusions: These findings further support a cellular recessive mechanism for cystogenesis in ADPKD primarily caused by inactivating germline and somatic variants of PKD1 or PKD2 genes in kidney cyst epithelium.
Trial registration: ClinicalTrials.gov NCT00792155.
Copyright © 2021 by the American Society of Nephrology.
Figures


References
-
- Kidney Disease Improving Global Outcomes (KDIGO) . Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Available at https://www.kidney-international.org/action/showPdf?pii=S2157-1716%2815%.... Accessed November 5, 2021 - PMC - PubMed
-
- Heyer CM, Sundsbak JL, Abebe KZ, Chapman AB, Torres VE, Grantham JJ, et al. ; HALT PKD and CRISP Investigators : Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 2872–2884, 2016 - PMC - PubMed
-
- Cornec-Le Gall E, Audrézet MP, Le Meur Y, Chen JM, Férec C: Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum Mutat 35: 1393–1406, 2014 - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
