

SARS-CoV-2 evolution in animals suggests mechanisms for rapid variant selection
- PMID: 34716263
- PMCID: PMC8612357
- DOI: 10.1073/pnas.2105253118
SARS-CoV-2 evolution in animals suggests mechanisms for rapid variant selection
Abstract
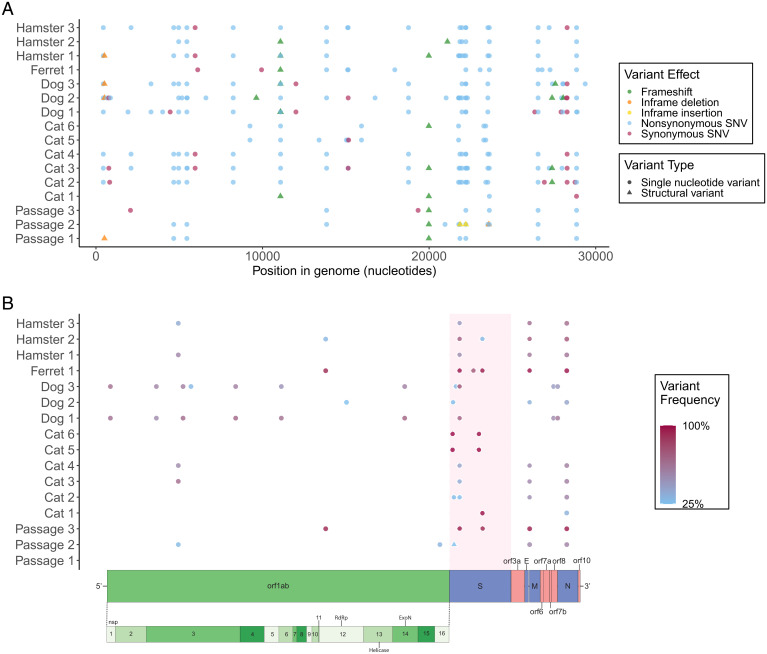
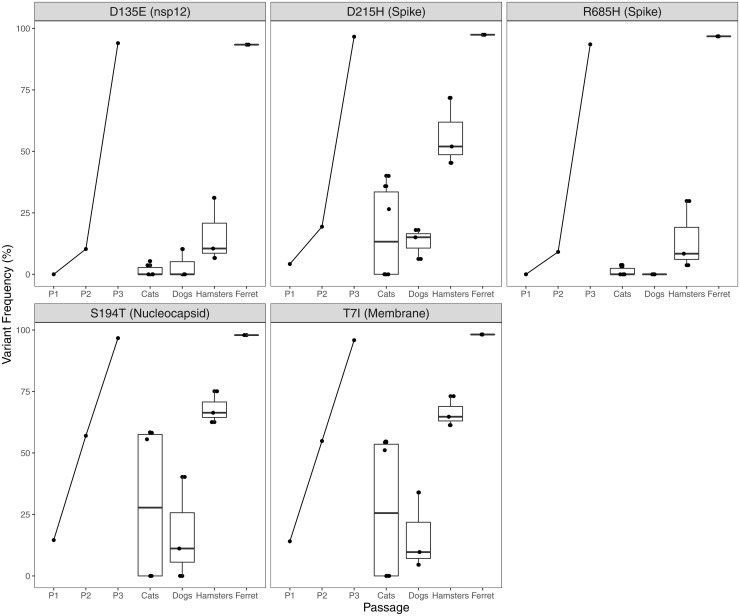
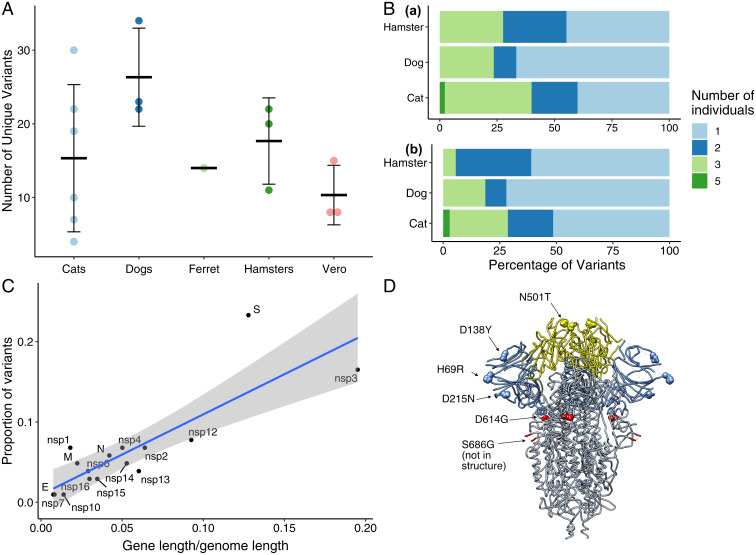
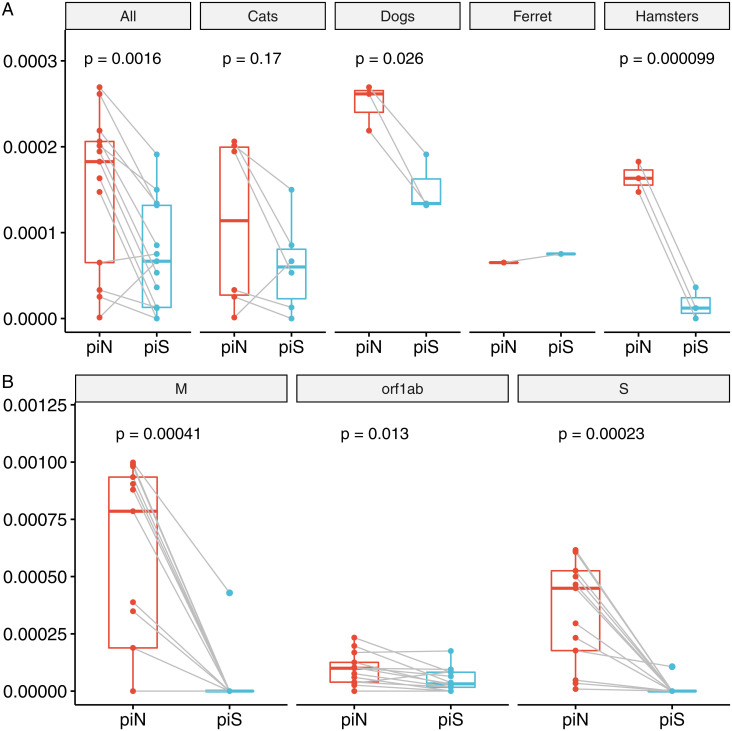
SARS-CoV-2 spillback from humans into domestic and wild animals has been well documented, and an accumulating number of studies illustrate that human-to-animal transmission is widespread in cats, mink, deer, and other species. Experimental inoculations of cats, mink, and ferrets have perpetuated transmission cycles. We sequenced full genomes of Vero cell-expanded SARS-CoV-2 inoculum and viruses recovered from cats (n = 6), dogs (n = 3), hamsters (n = 3), and a ferret (n = 1) following experimental exposure. Five nonsynonymous changes relative to the USA-WA1/2020 prototype strain were near fixation in the stock used for inoculation but had reverted to wild-type sequences at these sites in dogs, cats, and hamsters within 1- to 3-d postexposure. A total of 14 emergent variants (six in nonstructural genes, six in spike, and one each in orf8 and nucleocapsid) were detected in viruses recovered from animals. This included substitutions in spike residues H69, N501, and D614, which also vary in human lineages of concern. Even though a live virus was not cultured from dogs, substitutions in replicase genes were detected in amplified sequences. The rapid selection of SARS-CoV-2 variants in vitro and in vivo reveals residues with functional significance during host switching. These observations also illustrate the potential for spillback from animal hosts to accelerate the evolution of new viral lineages, findings of particular concern for dogs and cats living in households with COVID-19 patients. More generally, this glimpse into viral host switching reveals the unrealized rapidity and plasticity of viral evolution in experimental animal model systems.
Keywords: SARS-CoV-2; companion animals; host adaptation; spillover; viral variants.
Conflict of interest statement
The authors declare no competing interest.
Figures
Update of
-
SARS-CoV-2 evolution in animals suggests mechanisms for rapid variant selection.bioRxiv [Preprint]. 2021 Mar 9:2021.03.05.434135. doi: 10.1101/2021.03.05.434135. bioRxiv. 2021. Update in: Proc Natl Acad Sci U S A. 2021 Nov 2;118(44):e2105253118. doi: 10.1073/pnas.2105253118. PMID: 33758844 Free PMC article. Updated. Preprint.
Comment in
-
Data analysis of viral complementary DNA/RNA sequences for low-frequency intrahost-single nucleotide variants in COVID-19.Proc Natl Acad Sci U S A. 2022 Aug 23;119(34):e2205269119. doi: 10.1073/pnas.2205269119. Epub 2022 Aug 1. Proc Natl Acad Sci U S A. 2022. PMID: 35914186 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
