

Multiplexed single-cell proteomics using SCoPE2
- PMID: 34716448
- PMCID: PMC8643348
- DOI: 10.1038/s41596-021-00616-z
Multiplexed single-cell proteomics using SCoPE2
Abstract
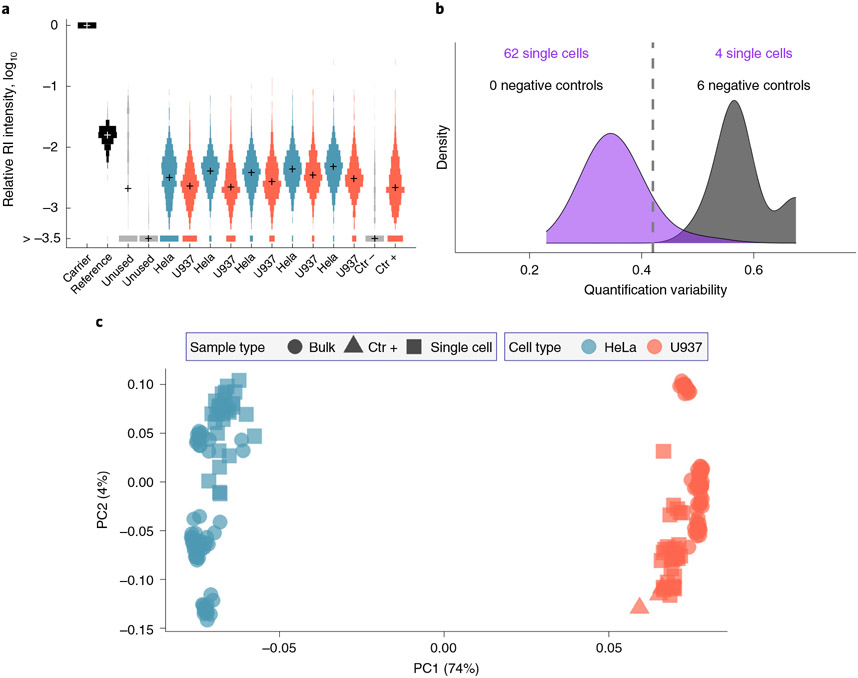
Many biological systems are composed of diverse single cells. This diversity necessitates functional and molecular single-cell analysis. Single-cell protein analysis has long relied on affinity reagents, but emerging mass-spectrometry methods (either label-free or multiplexed) have enabled quantifying >1,000 proteins per cell while simultaneously increasing the specificity of protein quantification. Here we describe the Single Cell ProtEomics (SCoPE2) protocol, which uses an isobaric carrier to enhance peptide sequence identification. Single cells are isolated by FACS or CellenONE into multiwell plates and lysed by Minimal ProteOmic sample Preparation (mPOP), and their peptides labeled by isobaric mass tags (TMT or TMTpro) for multiplexed analysis. SCoPE2 affords a cost-effective single-cell protein quantification that can be fully automated using widely available equipment and scaled to thousands of single cells. SCoPE2 uses inexpensive reagents and is applicable to any sample that can be processed to a single-cell suspension. The SCoPE2 workflow allows analyzing ~200 single cells per 24 h using only standard commercial equipment. We emphasize experimental steps and benchmarks required for achieving quantitative protein analysis.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
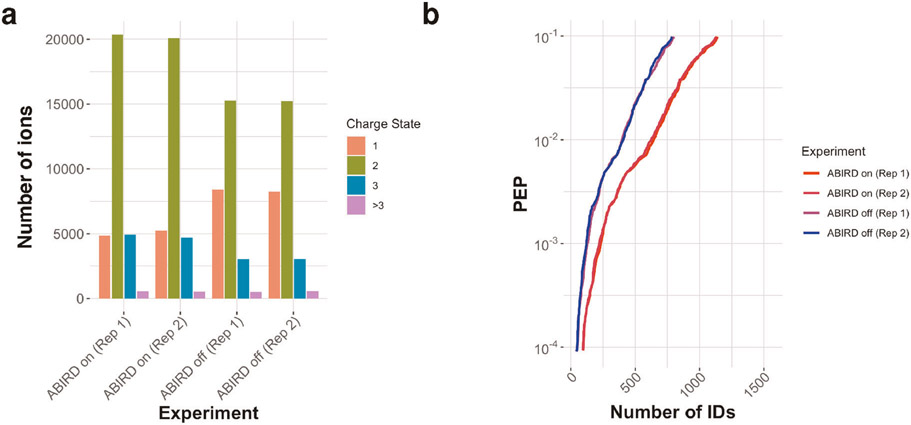
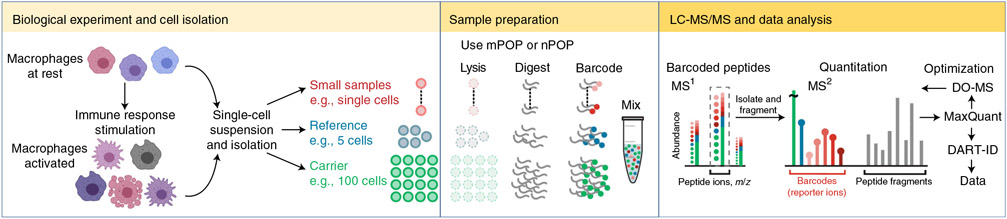
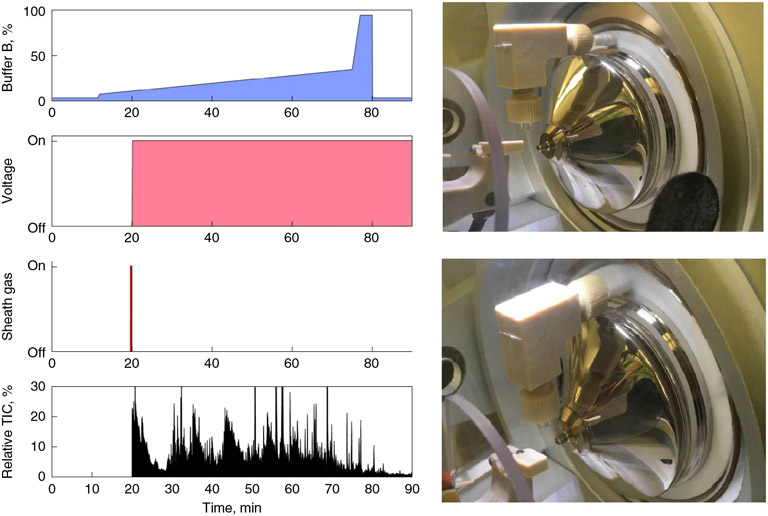
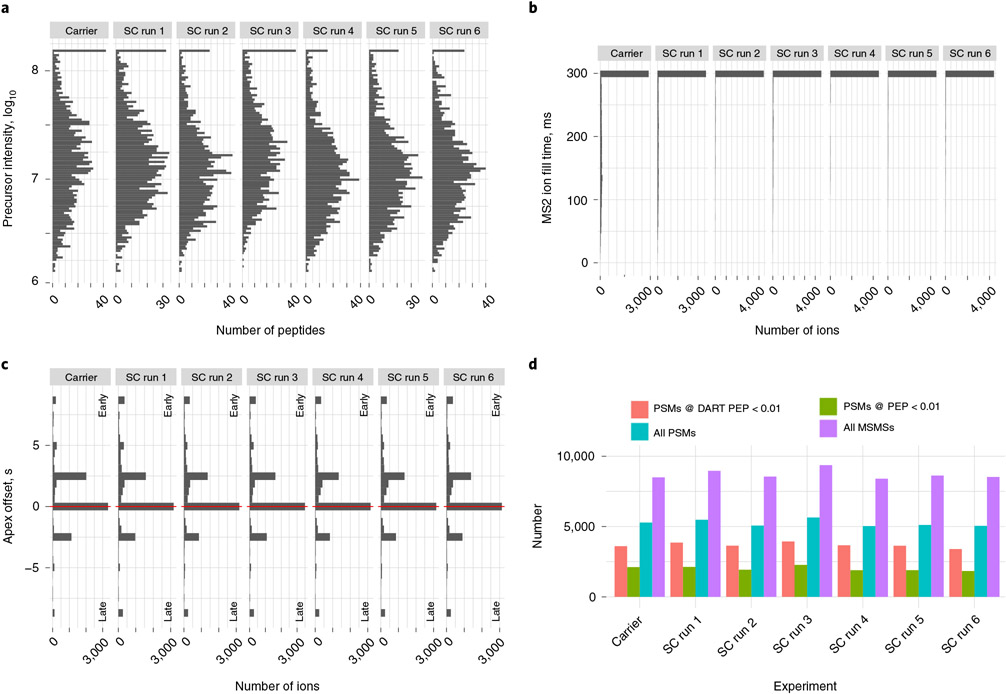
References
Related links
-
- Budnik B et al. Genome Biol. 19, 161 (2018): https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1547-5 - DOI - PMC - PubMed
-
- Specht H & Slavov NJ Proteome Res. 20, 880887 (2021): https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00675 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
