

Distance-based reconstruction of protein quaternary structures from inter-chain contacts
- PMID: 34716620
- PMCID: PMC8816881
- DOI: 10.1002/prot.26269
Distance-based reconstruction of protein quaternary structures from inter-chain contacts
Abstract
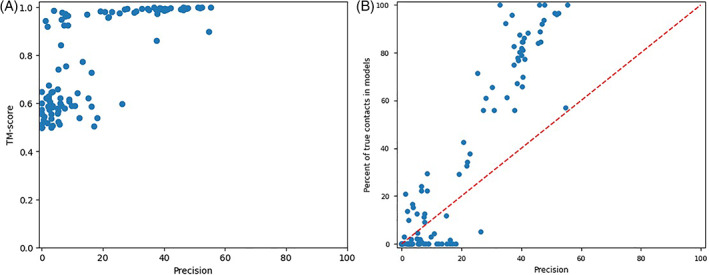
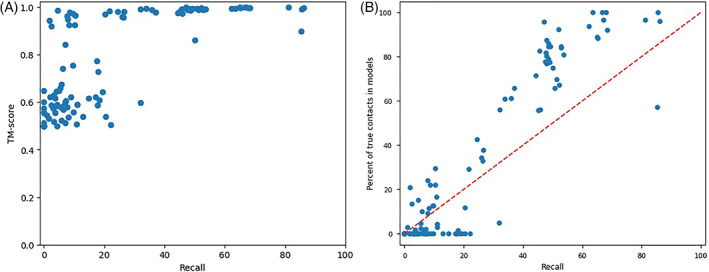
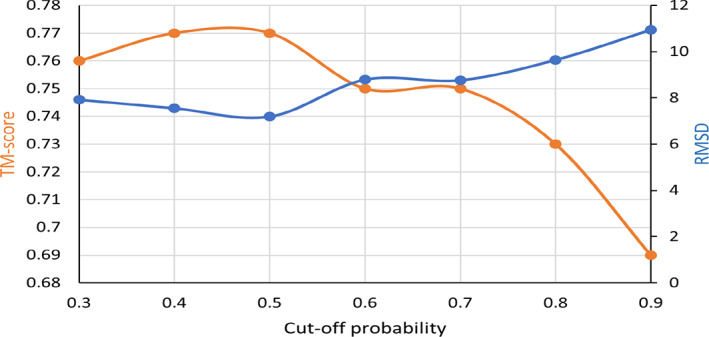
Predicting the quaternary structure of protein complex is an important problem. Inter-chain residue-residue contact prediction can provide useful information to guide the ab initio reconstruction of quaternary structures. However, few methods have been developed to build quaternary structures from predicted inter-chain contacts. Here, we develop the first method based on gradient descent optimization (GD) to build quaternary structures of protein dimers utilizing inter-chain contacts as distance restraints. We evaluate GD on several datasets of homodimers and heterodimers using true/predicted contacts and monomer structures as input. GD consistently performs better than both simulated annealing and Markov Chain Monte Carlo simulation. Starting from an arbitrarily quaternary structure randomly initialized from the tertiary structures of protein chains and using true inter-chain contacts as input, GD can reconstruct high-quality structural models for homodimers and heterodimers with average TM-score ranging from 0.92 to 0.99 and average interface root mean square distance from 0.72 Å to 1.64 Å. On a dataset of 115 homodimers, using predicted inter-chain contacts as restraints, the average TM-score of the structural models built by GD is 0.76. For 46% of the homodimers, high-quality structural models with TM-score ≥ 0.9 are reconstructed from predicted contacts. There is a strong correlation between the quality of the reconstructed models and the precision and recall of predicted contacts. Only a moderate precision or recall of inter-chain contact prediction is needed to build good structural models for most homodimers. Moreover, GD improves the quality of quaternary structures predicted by AlphaFold2 on a Critical Assessment of Techniques for Protein Structure Prediction-Critical Assessments of Predictions of Interactions dataset.
Keywords: distance-based modeling; gradient descent optimization; inter-chain contact prediction; protein complex; protein quaternary structure modeling.
© 2021 The Authors. Proteins: Structure, Function, and Bioinformatics published by Wiley Periodicals LLC.
Figures
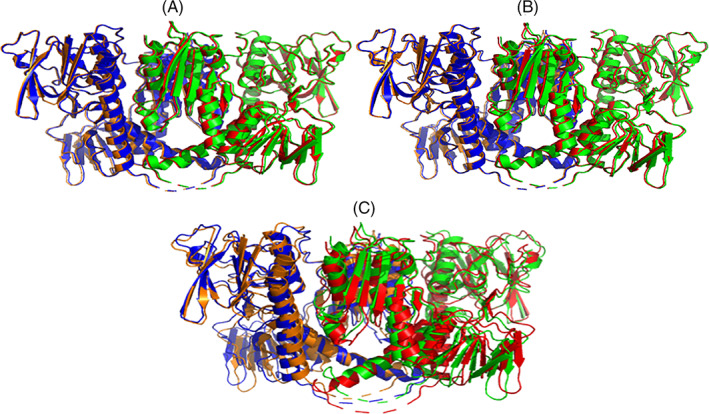
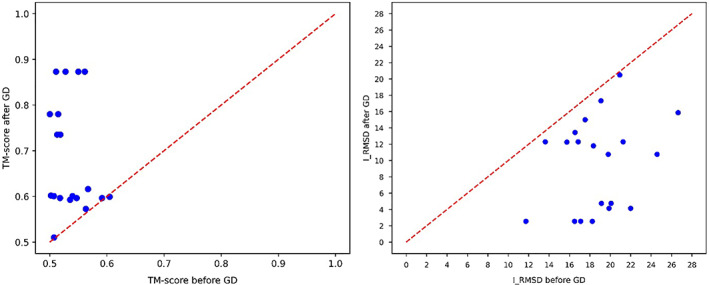
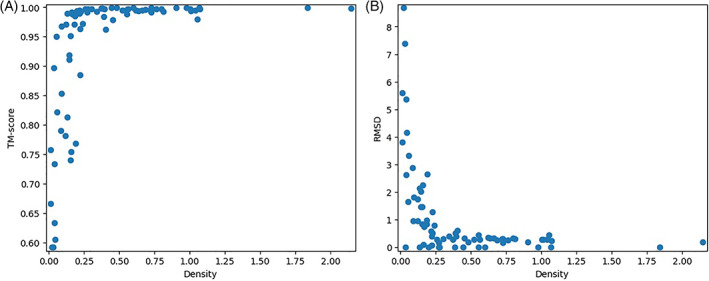
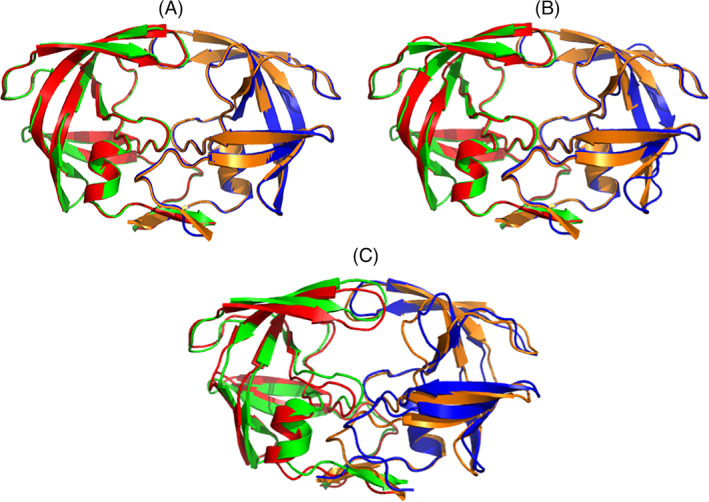
References
-
- Hadarovich A, Kalinouski A, Tuzikov AV. Deep learning approach with rotate‐shift invariant input to predict protein homodimer structure. Bioinformatics Research and Applications. Springer; 2020:296‐303.
-
- Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein−protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731‐1737. - PubMed
-
- Chen R, Li L, Weng Z. ZDOCK: an initial‐stage protein‐docking algorithm. Proteins. 2003;52:80‐87. - PubMed
-
- Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45‐50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
