

Developmental Programming: Prenatal Testosterone Excess on Liver and Muscle Coding and Noncoding RNA in Female Sheep
- PMID: 34718504
- PMCID: PMC8667859
- DOI: 10.1210/endocr/bqab225
Developmental Programming: Prenatal Testosterone Excess on Liver and Muscle Coding and Noncoding RNA in Female Sheep
Abstract
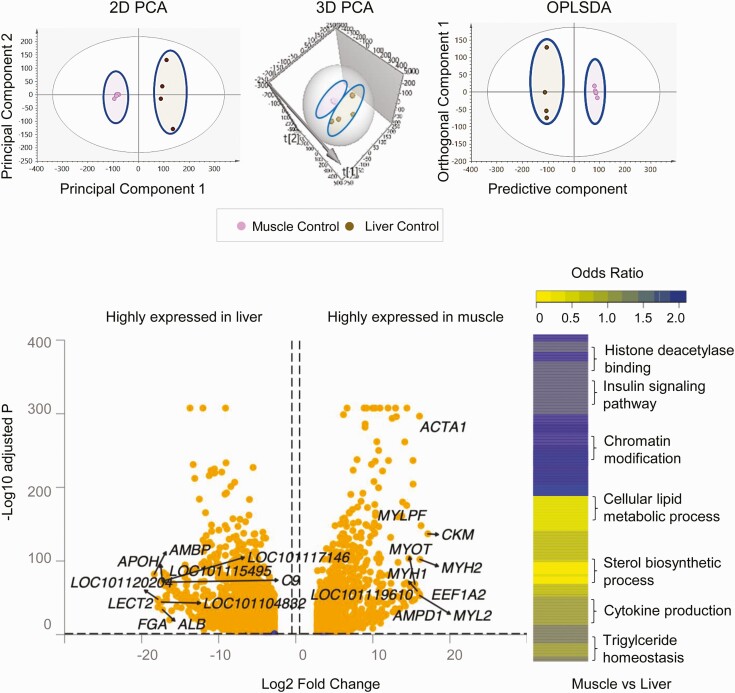
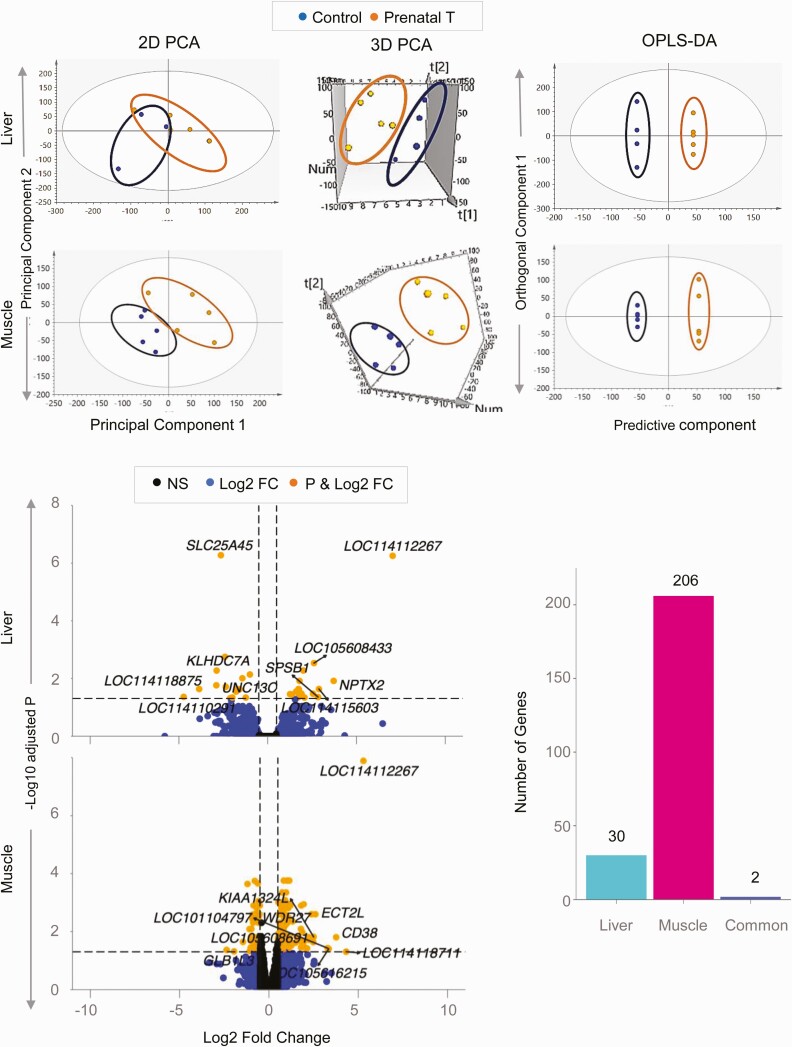
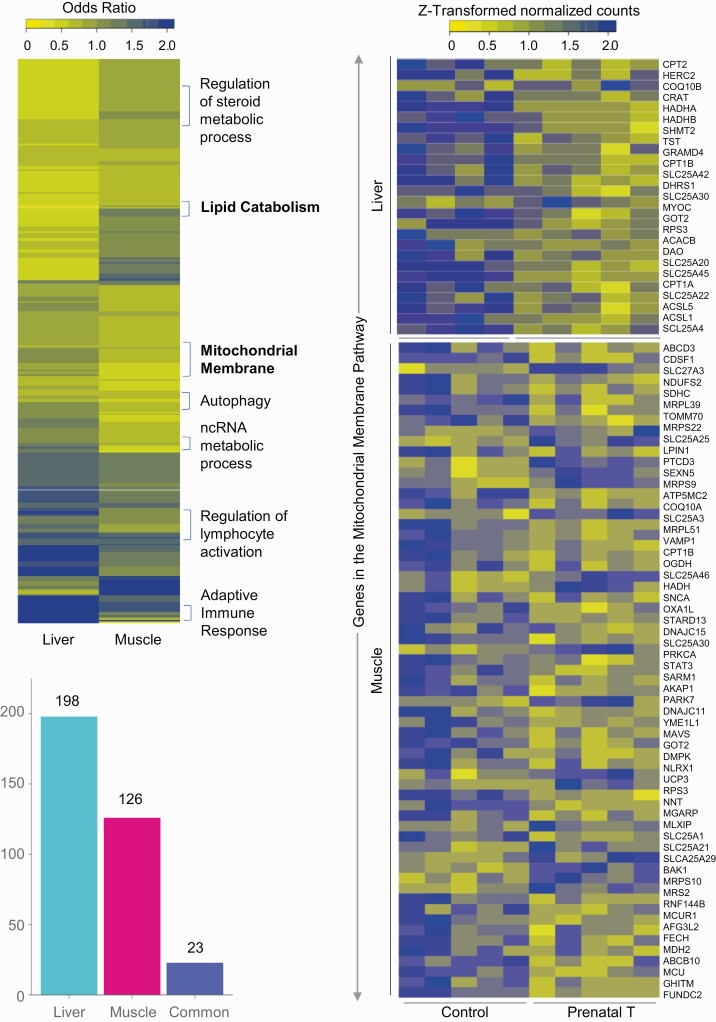
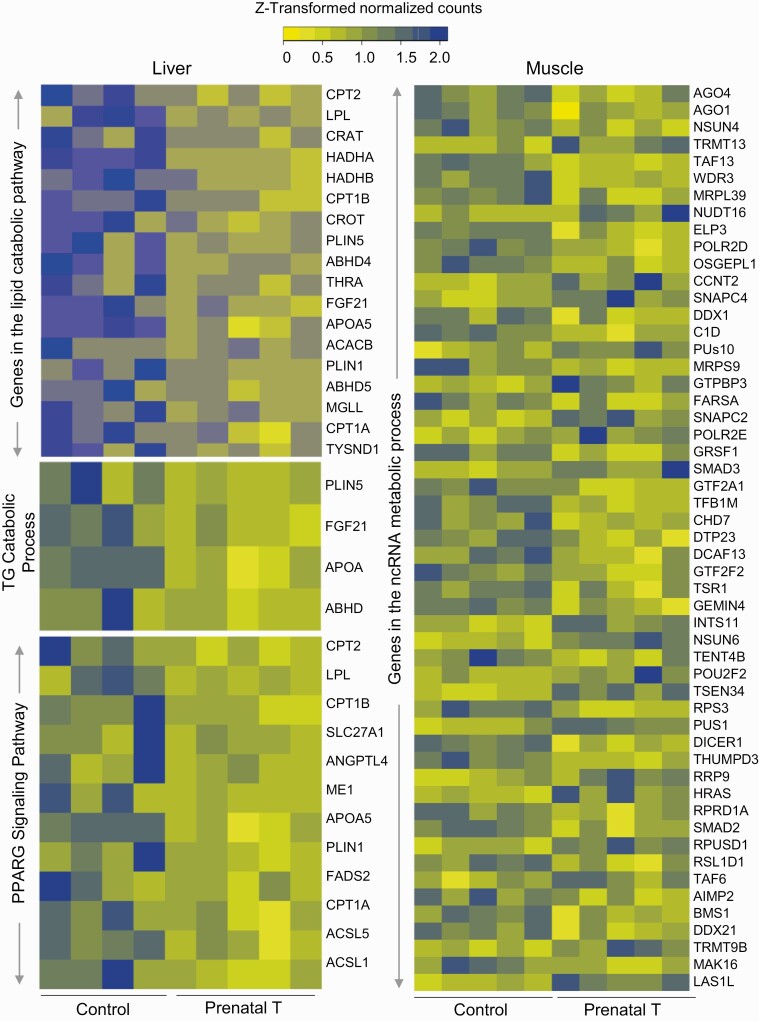
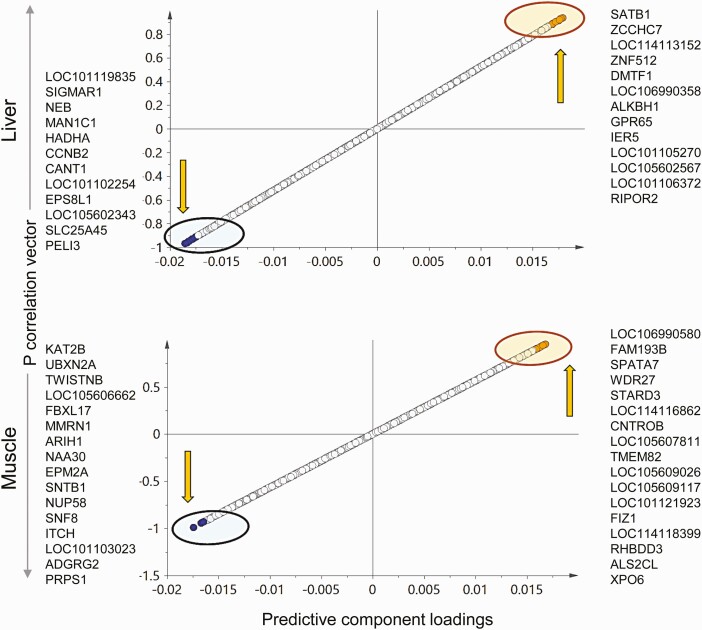
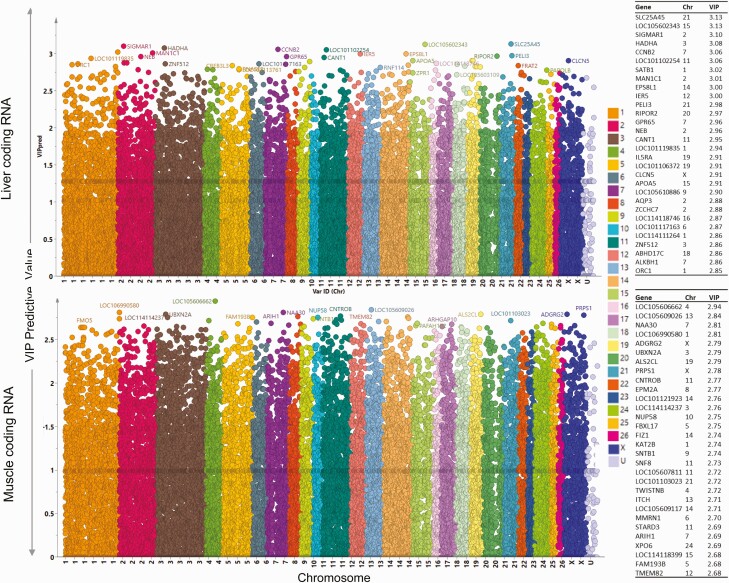
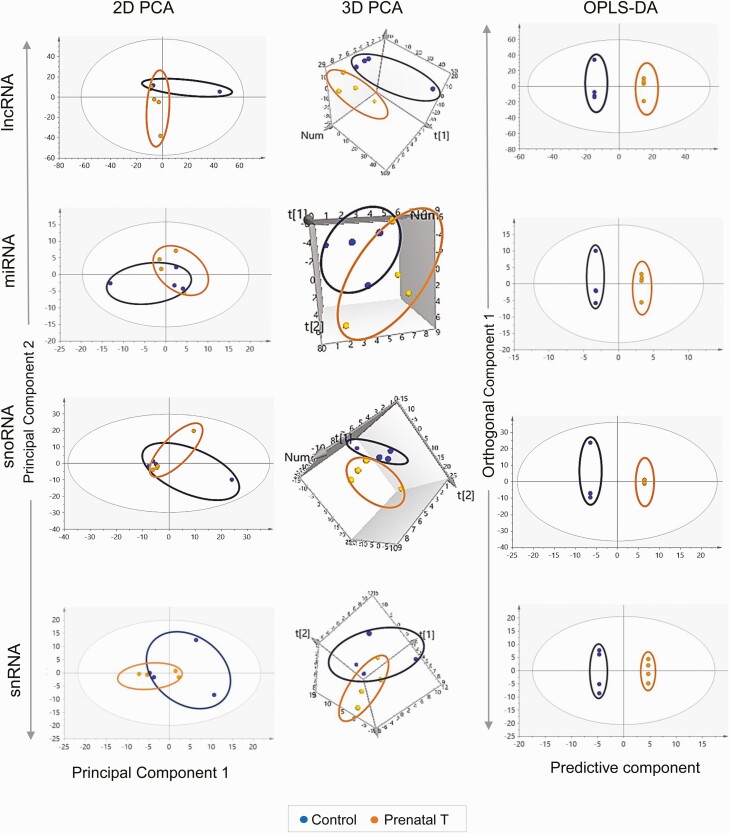
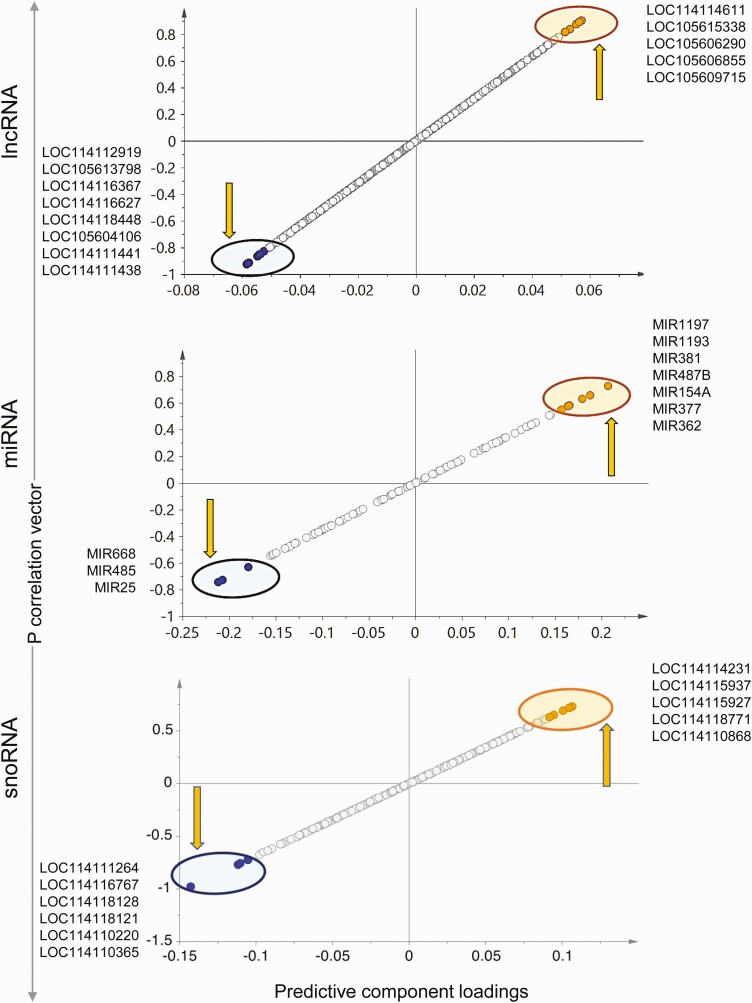
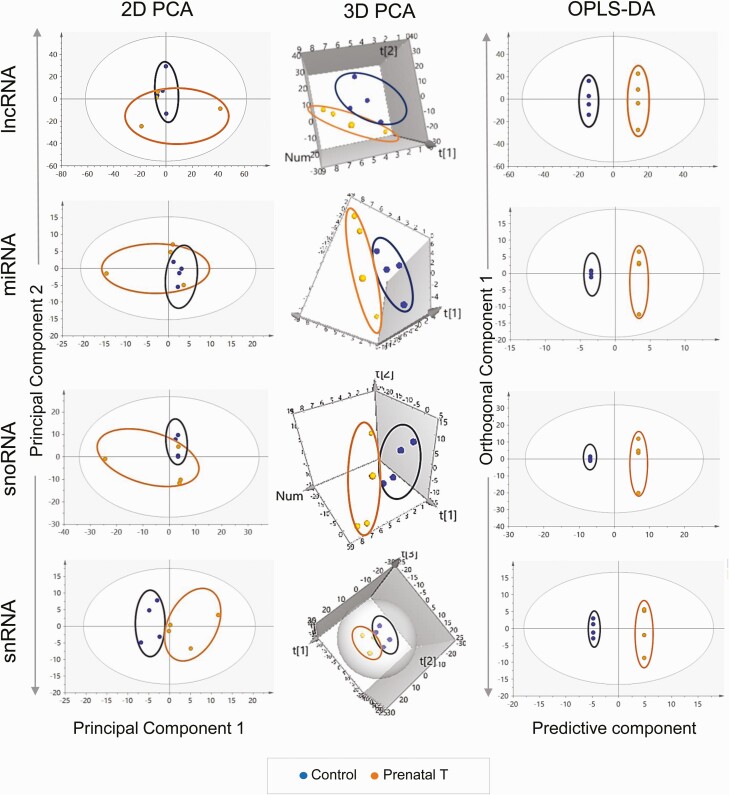
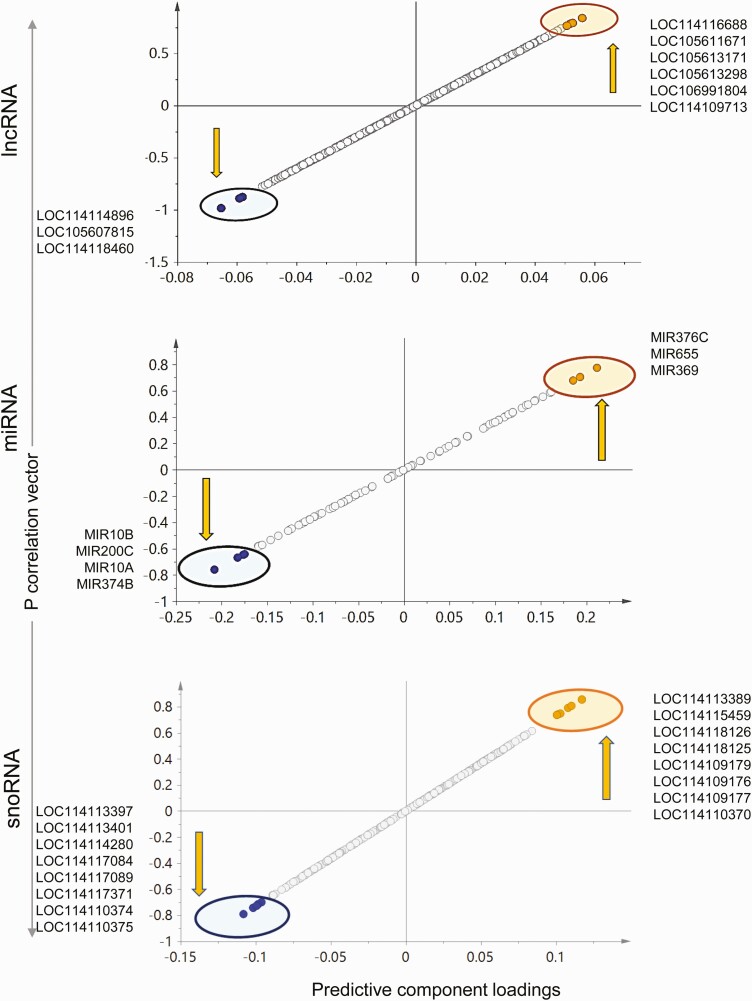
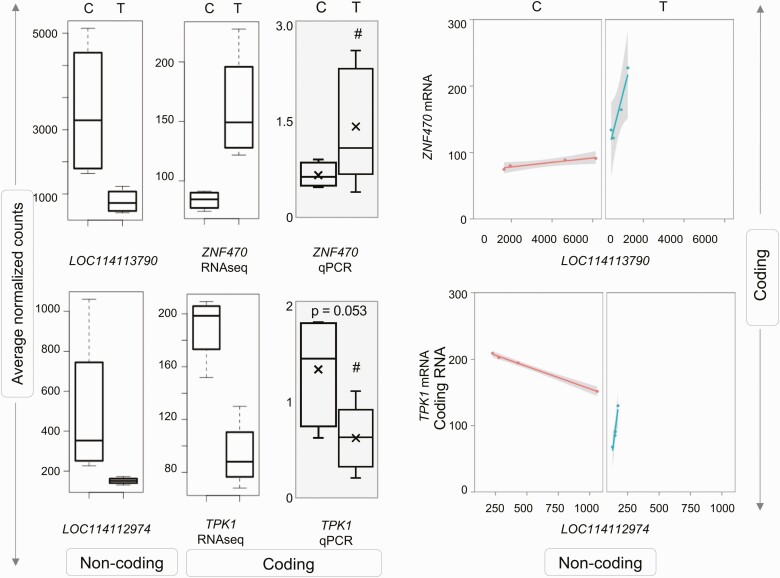
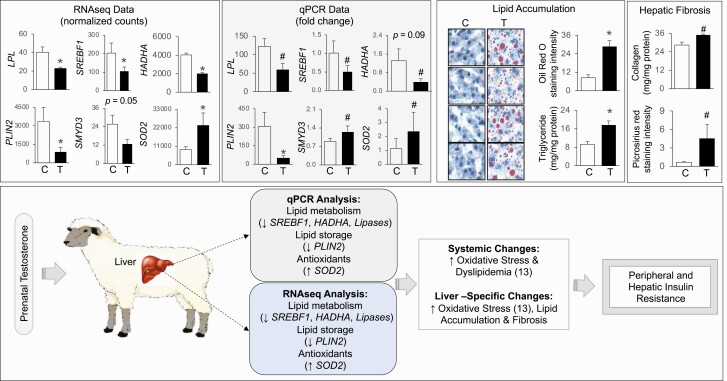
Prenatal testosterone (T)-treated female sheep manifest peripheral insulin resistance, ectopic lipid accumulation, and insulin signaling disruption in liver and muscle. This study investigated transcriptional changes and transcriptome signature of prenatal T excess-induced hepatic and muscle-specific metabolic disruptions. Genome-wide coding and noncoding (nc) RNA expression in liver and muscle from 21-month-old prenatal T-treated (T propionate 100 mg intramuscular twice weekly from days 30-90 of gestation; term: 147 days) and control females were compared. Prenatal T (1) induced differential expression of messenger RNAs (mRNAs) in liver (15 down, 17 up) and muscle (66 down, 176 up) (false discovery rate < 0.05, absolute log2 fold change > 0.5); (2) downregulated mitochondrial pathway genes in liver and muscle; (3) downregulated hepatic lipid catabolism and peroxisome proliferator-activated receptor (PPAR) signaling gene pathways; (4) modulated noncoding RNA (ncRNA) metabolic processes gene pathway in muscle; and (5) downregulated 5 uncharacterized long noncoding RNA (lncRNA) in the muscle but no ncRNA changes in the liver. Correlation analysis showed downregulation of lncRNAs LOC114112974 and LOC105607806 was associated with decreased TPK1, and LOC114113790 with increased ZNF470 expression. Orthogonal projections to latent structures discriminant analysis identified mRNAs HADHA and SLC25A45, and microRNAs MIR154A, MIR25, and MIR487B in the liver and ARIH1 and ITCH and miRNAs MIR369, MIR10A, and MIR10B in muscle as potential biomarkers of prenatal T excess. These findings suggest downregulation of mitochondria, lipid catabolism, and PPAR signaling genes in the liver and dysregulation of mitochondrial and ncRNA gene pathways in muscle are contributors of lipotoxic and insulin-resistant hepatic and muscle phenotype. Gestational T excess programming of metabolic dysfunctions involve tissue-specific ncRNA-modulated transcriptional changes.
Keywords: DOHAD; RNA sequencing; metabolic tissues; ovine.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
Similar articles
-
Developmental programming: Impact of prenatal bisphenol-A exposure on liver and muscle transcriptome of female sheep.Toxicol Appl Pharmacol. 2022 Sep 15;451:116161. doi: 10.1016/j.taap.2022.116161. Epub 2022 Jul 8. Toxicol Appl Pharmacol. 2022. PMID: 35817127 Free PMC article.
-
Developmental programming: adverse sexually dimorphic transcriptional programming of gestational testosterone excess in cardiac left ventricle of fetal sheep.Sci Rep. 2023 Feb 15;13(1):2682. doi: 10.1038/s41598-023-29212-9. Sci Rep. 2023. PMID: 36792653 Free PMC article.
-
Developmental programming: Metabolic tissue-specific changes in endoplasmic reticulum stress, mitochondrial oxidative and telomere length status induced by prenatal testosterone excess in the female sheep.Mol Cell Endocrinol. 2021 Apr 15;526:111207. doi: 10.1016/j.mce.2021.111207. Epub 2021 Feb 17. Mol Cell Endocrinol. 2021. PMID: 33607270 Free PMC article.
-
Non-Coding RNAs as Potential Novel Biomarkers for Early Diagnosis of Hepatic Insulin Resistance.Int J Mol Sci. 2020 Jun 11;21(11):4182. doi: 10.3390/ijms21114182. Int J Mol Sci. 2020. PMID: 32545342 Free PMC article. Review.
-
Prenatal Testosterone Programming of Insulin Resistance in the Female Sheep.Adv Exp Med Biol. 2017;1043:575-596. doi: 10.1007/978-3-319-70178-3_25. Adv Exp Med Biol. 2017. PMID: 29224111 Review.
Cited by
-
Developmental programming: Impact of prenatal bisphenol-A exposure on liver and muscle transcriptome of female sheep.Toxicol Appl Pharmacol. 2022 Sep 15;451:116161. doi: 10.1016/j.taap.2022.116161. Epub 2022 Jul 8. Toxicol Appl Pharmacol. 2022. PMID: 35817127 Free PMC article.
-
Exploratory analysis of differences at the transcriptional interface between the maternal and fetal compartments of the sheep placenta and potential influence of fetal sex.Mol Cell Endocrinol. 2025 Jun 1;603:112546. doi: 10.1016/j.mce.2025.112546. Epub 2025 Apr 12. Mol Cell Endocrinol. 2025. PMID: 40222550
-
Developmental programming: adverse sexually dimorphic transcriptional programming of gestational testosterone excess in cardiac left ventricle of fetal sheep.Sci Rep. 2023 Feb 15;13(1):2682. doi: 10.1038/s41598-023-29212-9. Sci Rep. 2023. PMID: 36792653 Free PMC article.
-
Maternal plasma microRNAs as potential biomarkers for triaging pregnancies of unknown location and ectopic pregnancy diagnosis.Noncoding RNA Res. 2025 May 8;13:162-173. doi: 10.1016/j.ncrna.2025.05.005. eCollection 2025 Aug. Noncoding RNA Res. 2025. PMID: 40501483 Free PMC article.
-
Developmental programming: Sex-specific effects of prenatal exposure to a real-life mixture of environmental chemicals on liver function and transcriptome in sheep.Environ Pollut. 2025 Feb 15;367:125630. doi: 10.1016/j.envpol.2025.125630. Epub 2025 Jan 3. Environ Pollut. 2025. PMID: 39756566
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
