

Progressive fibrosing interstitial lung disease: prevalence and clinical outcome
- PMID: 34719401
- PMCID: PMC8559348
- DOI: 10.1186/s12931-021-01879-6
Progressive fibrosing interstitial lung disease: prevalence and clinical outcome
Abstract
Background: The progressive fibrosing (PF) phenotype of interstitial lung disease (ILD) is characterised by worsening respiratory symptoms, lung function, and extent of fibrosis on high-resolution computed tomography. We aimed to investigate the prevalence and clinical outcomes of PF-ILD in a real-world cohort and assess the prognostic significance of the PF-ILD diagnostic criteria.
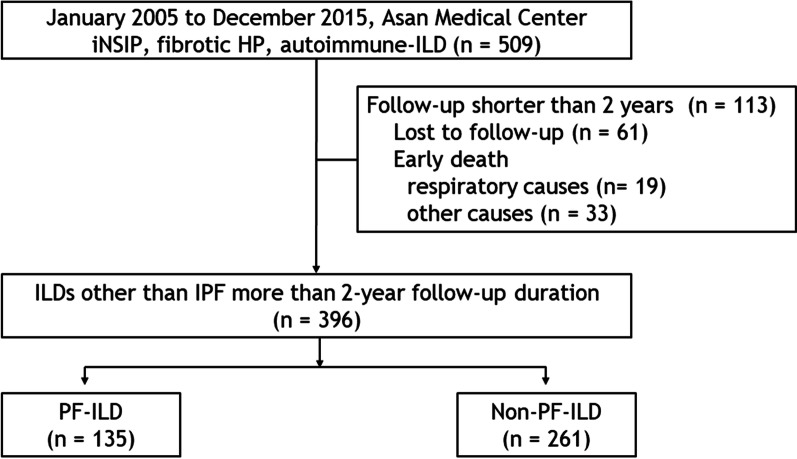
Methods: Clinical data of patients with fibrosing ILD other than idiopathic pulmonary fibrosis (IPF) consecutively diagnosed at a single centre were retrospectively reviewed. A PF phenotype was defined based on the criteria used in the INBUILD trial.
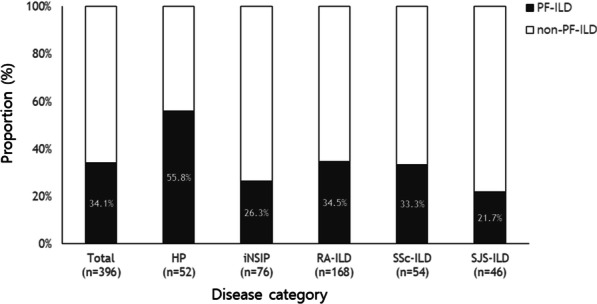
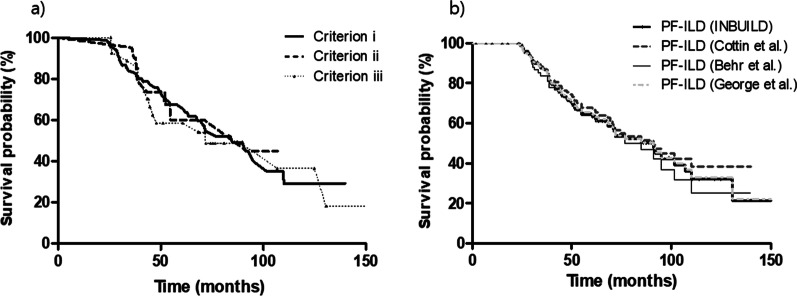
Results: The median follow-up duration was 62.7 months. Of the total of 396 patients, the mean age was 58.1 years, 39.9% were men, and rheumatoid arthritis-ILD was the most common (42.4%). A PF phenotype was identified in 135 patients (34.1%). The PF-ILD group showed lower forced vital capacity and total lung capacity (TLC) than the non-PF-ILD group. The PF-ILD group also showed poorer survival (median survival, 91.2 months vs. not reached; P < 0.001) than the non-PF-ILD group. In multivariable Cox analysis adjusted for age, DLCO, HRCT pattern, and specific diagnosis, PF phenotype was independent prognostic factor (hazard ratio, 3.053; P < 0.001) in patients with fibrosing ILD. Each criterion of PF-ILD showed similar survival outcomes.
Conclusions: Our results showed that approximately 34% of patients with non-IPF fibrosing ILD showed a progressive phenotype and a poor outcome similar to that of IPF, regardless of the diagnostic criteria used.
Keywords: Interstitial lung disease; Outcome; Phenotype; Prevalence; Progressive.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. - DOI - PMC - PubMed
-
- Wijsenbeek M, Kreuter M, Olson A, Fischer A, Bendstrup E, Wells CD, Denton CP, Mounir B, Zouad-Lejour L, Quaresma M. Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin. 2019;35:2015–2024. doi: 10.1080/03007995.2019.1647040. - DOI - PubMed
-
- Ley B, Newton CA, Arnould I, Elicker BM, Henry TS, Vittinghoff E, Golden JA, Jones KD, Batra K, Torrealba J, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5:639–647. doi: 10.1016/S2213-2600(17)30216-3. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
