

Timing RNA polymerase pausing with TV-PRO-seq
- PMID: 34723238
- PMCID: PMC8547241
- DOI: 10.1016/j.crmeth.2021.100083
Timing RNA polymerase pausing with TV-PRO-seq
Abstract
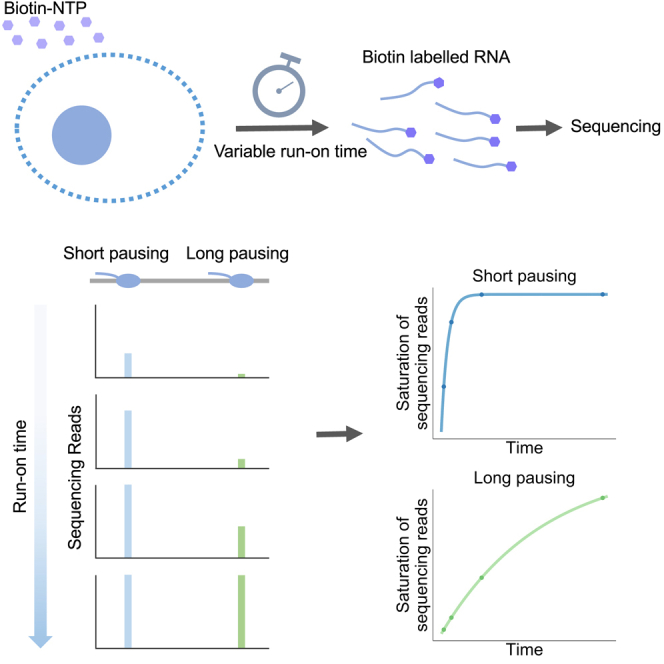
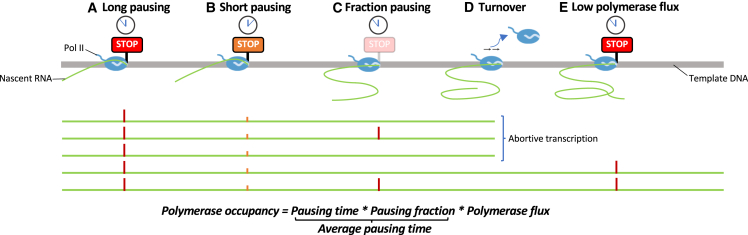
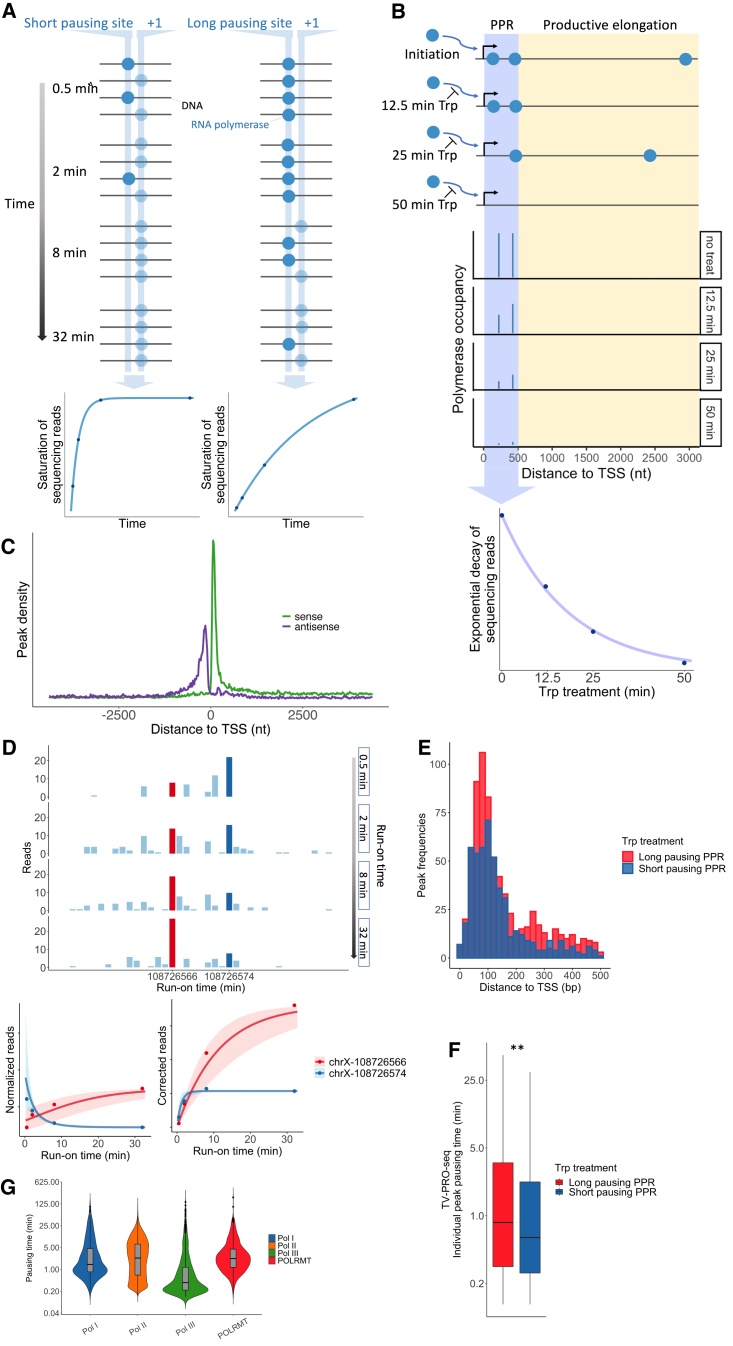
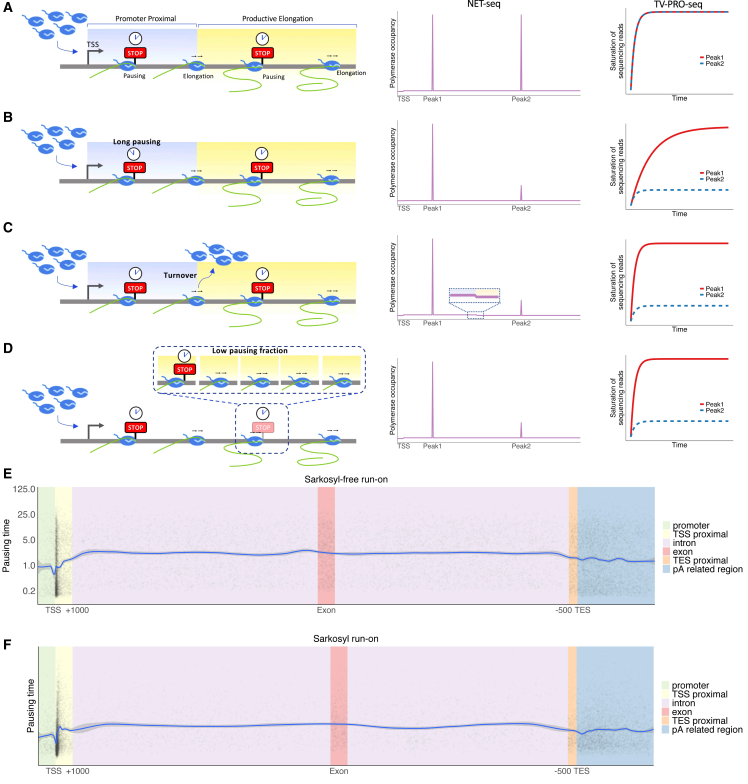
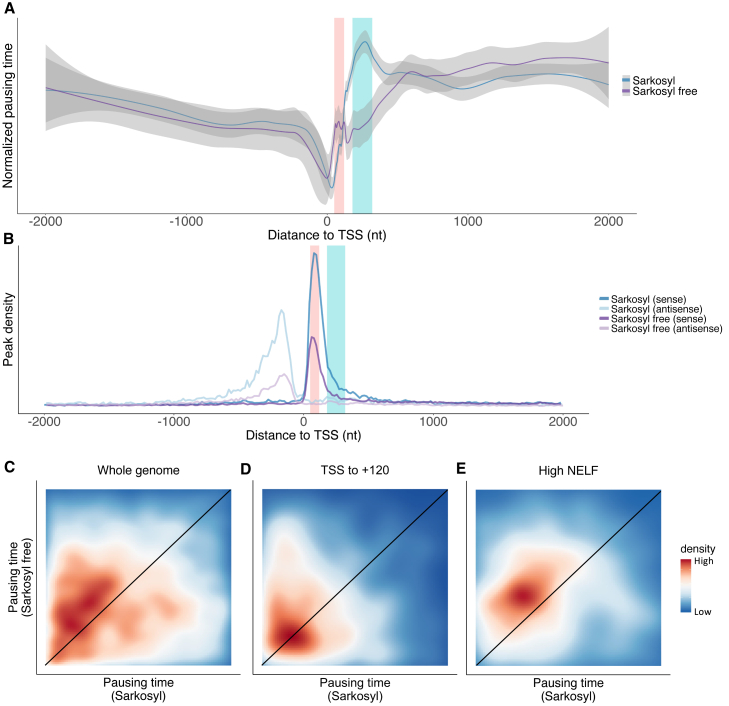
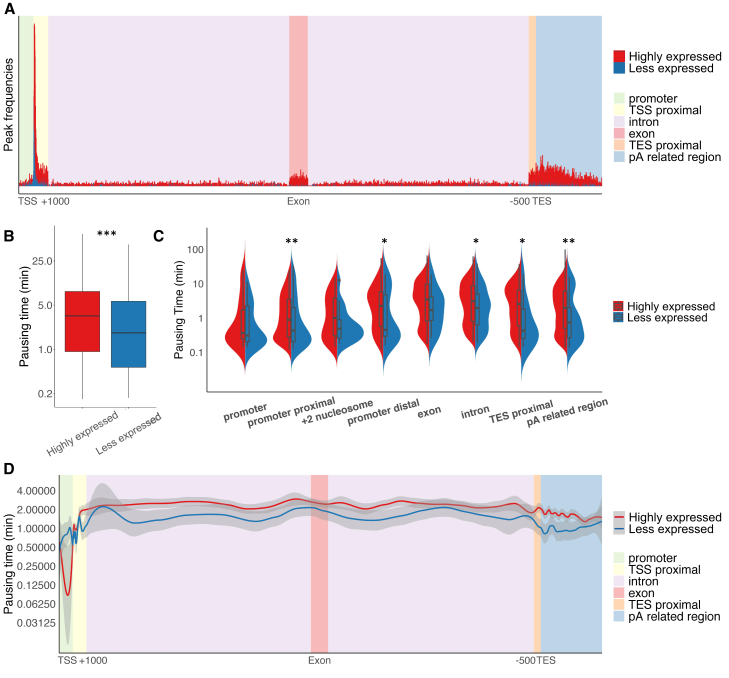
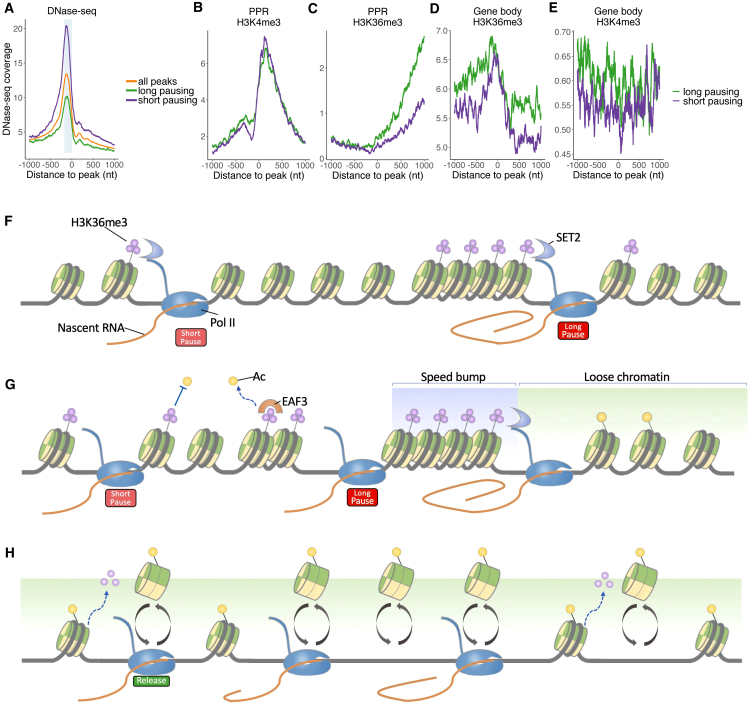
Transcription of many genes in metazoans is subject to polymerase pausing, which is the transient stop of transcriptionally engaged polymerases. This is known to mainly occur in promoter-proximal regions but it is not well understood. In particular, a genome-wide measurement of pausing times at high resolution has been lacking. We present here the time-variant precision nuclear run-on and sequencing (TV-PRO-seq) assay, an extension of the standard PRO-seq that allows us to estimate genome-wide pausing times at single-base resolution. Its application to human cells demonstrates that, proximal to promoters, polymerases pause more frequently but for shorter times than in other genomic regions. Comparison with single-cell gene expression data reveals that the polymerase pausing times are longer in highly expressed genes, while transcriptionally noisier genes have higher pausing frequencies and slightly longer pausing times. Analyses of histone modifications suggest that the marker H3K36me3 is related to the polymerase pausing.
Keywords: H3K36me3; NELF; PRO-seq; RNA polymerase; gene expression; next-generation sequencing; polymerase pausing; promoter-proximal pausing; transcription dynamics; transcriptional noise.
© 2021 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Al-Rfou R., Guillaume A., Almahairi A., Angermueller C., Bahdanau D., Ballas N., Bastien F., Bayer J., Belikov A., Belopolsky A., et al. arXiv; 2016. Theano: A Python Framework for Fast Computation of Mathematical Expressions. 1605.02688.
-
- Angers-Loustau A., Petrillo M., Bengtsson-Palme J., Berendonk T., Blais B., Chan K.G., Coque T.M., Hammer P., Hess S., Kagkli D.M., et al. The challenges of designing a benchmark strategy for bioinformatics pipelines in the identification of antimicrobial resistance determinants using next generation sequencing technologies. F1000Res. 2018;7 ISCB Comm J-459. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
