

The automated optimisation of a coarse-grained force field using free energy data
- PMID: 34723311
- PMCID: PMC8579472
- DOI: 10.1039/d0cp05041e
The automated optimisation of a coarse-grained force field using free energy data
Abstract
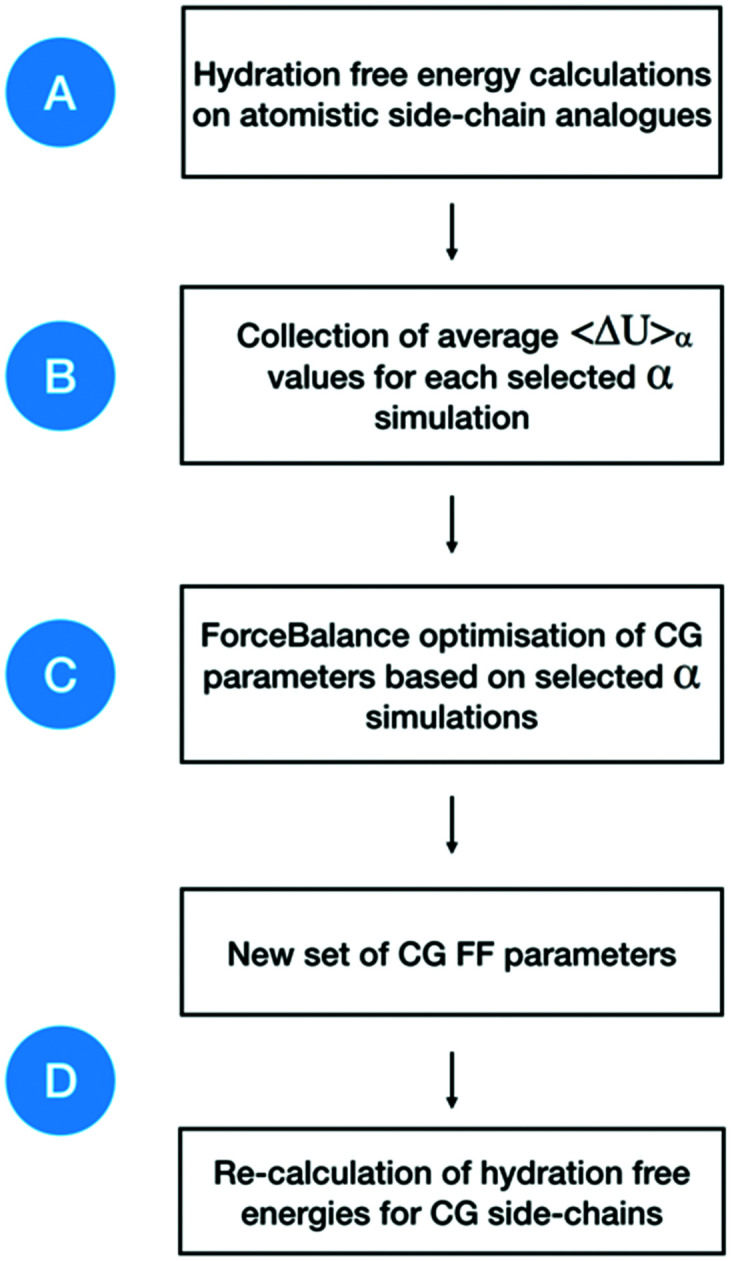
Atomistic models provide a detailed representation of molecular systems, but are sometimes inadequate for simulations of large systems over long timescales. Coarse-grained models enable accelerated simulations by reducing the number of degrees of freedom, at the cost of reduced accuracy. New optimisation processes to parameterise these models could improve their quality and range of applicability. We present an automated approach for the optimisation of coarse-grained force fields, by reproducing free energy data derived from atomistic molecular simulations. To illustrate the approach, we implemented hydration free energy gradients as a new target for force field optimisation in ForceBalance and applied it successfully to optimise the un-charged side-chains and the protein backbone in the SIRAH protein coarse-grain force field. The optimised parameters closely reproduced hydration free energies of atomistic models and gave improved agreement with experiment.
Conflict of interest statement
There are no conflicts to declare.
Figures

Similar articles
-
Coarse-grained force fields for molecular simulations.Methods Mol Biol. 2015;1215:125-49. doi: 10.1007/978-1-4939-1465-4_7. Methods Mol Biol. 2015. PMID: 25330962
-
Hybrid simulations: combining atomistic and coarse-grained force fields using virtual sites.Phys Chem Chem Phys. 2011 Jun 14;13(22):10437-48. doi: 10.1039/c0cp02981e. Epub 2011 Apr 15. Phys Chem Chem Phys. 2011. PMID: 21494747
-
Further optimization of a hybrid united-atom and coarse-grained force field for folding simulations: Improved backbone hydration and interactions between charged side chains.J Chem Theory Comput. 2012 Nov 13;8(11):4413-4424. doi: 10.1021/ct300696c. Epub 2012 Oct 11. J Chem Theory Comput. 2012. PMID: 23204949 Free PMC article.
-
The SIRAH force field: A suite for simulations of complex biological systems at the coarse-grained and multiscale levels.J Struct Biol. 2023 Sep;215(3):107985. doi: 10.1016/j.jsb.2023.107985. Epub 2023 Jun 16. J Struct Biol. 2023. PMID: 37331570 Review.
-
Coarse-grained force field: general folding theory.Phys Chem Chem Phys. 2011 Oct 14;13(38):16890-901. doi: 10.1039/c1cp20752k. Epub 2011 Jun 3. Phys Chem Chem Phys. 2011. PMID: 21643583 Free PMC article. Review.
Cited by
-
Biomolecular Condensates as Emerging Biomaterials: Functional Mechanisms and Advances in Computational and Experimental Approaches.Adv Mater. 2025 Sep;37(36):e10115. doi: 10.1002/adma.202510115. Epub 2025 Aug 13. Adv Mater. 2025. PMID: 40801528 Free PMC article. Review.
References
-
- Huggins D. J. Biggin P. C. Dämgen M. A. Essex J. W. Harris S. A. Henchman R. H. Khalid S. Kuzmanic A. Laughton C. A. Michel J. Mulholland A. J. Rosta E. Sansom M. S. P. van der Kamp M. W. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2019;9(3):e1393.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources