

Quantitative sequencing clarifies the role of disruptor taxa, oral microbiota, and strict anaerobes in the human small-intestine microbiome
- PMID: 34724979
- PMCID: PMC8561862
- DOI: 10.1186/s40168-021-01162-2
Quantitative sequencing clarifies the role of disruptor taxa, oral microbiota, and strict anaerobes in the human small-intestine microbiome
Abstract
Background: Upper gastrointestinal (GI) disorders and abdominal pain afflict between 12 and 30% of the worldwide population and research suggests these conditions are linked to the gut microbiome. Although large-intestine microbiota have been linked to several GI diseases, the microbiota of the human small intestine and its relation to human disease has been understudied. The small intestine is the major site for immune surveillance in the gut, and compared with the large intestine, it has greater than 100 times the surface area and a thinner and more permeable mucus layer.
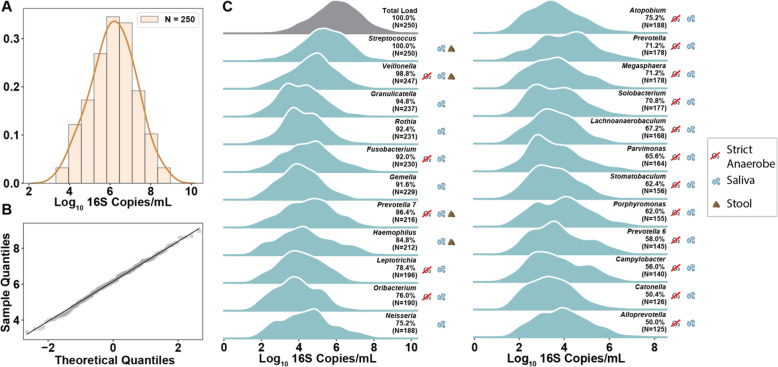
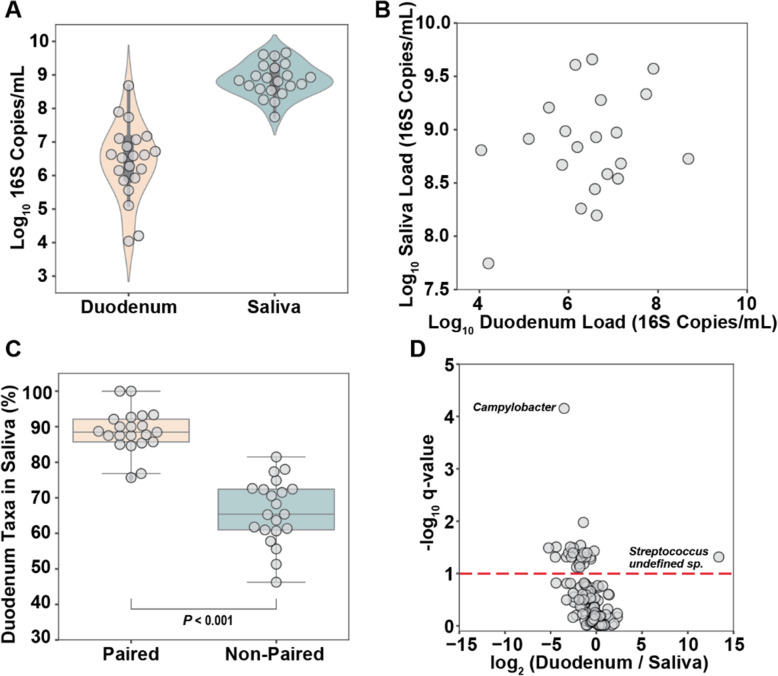
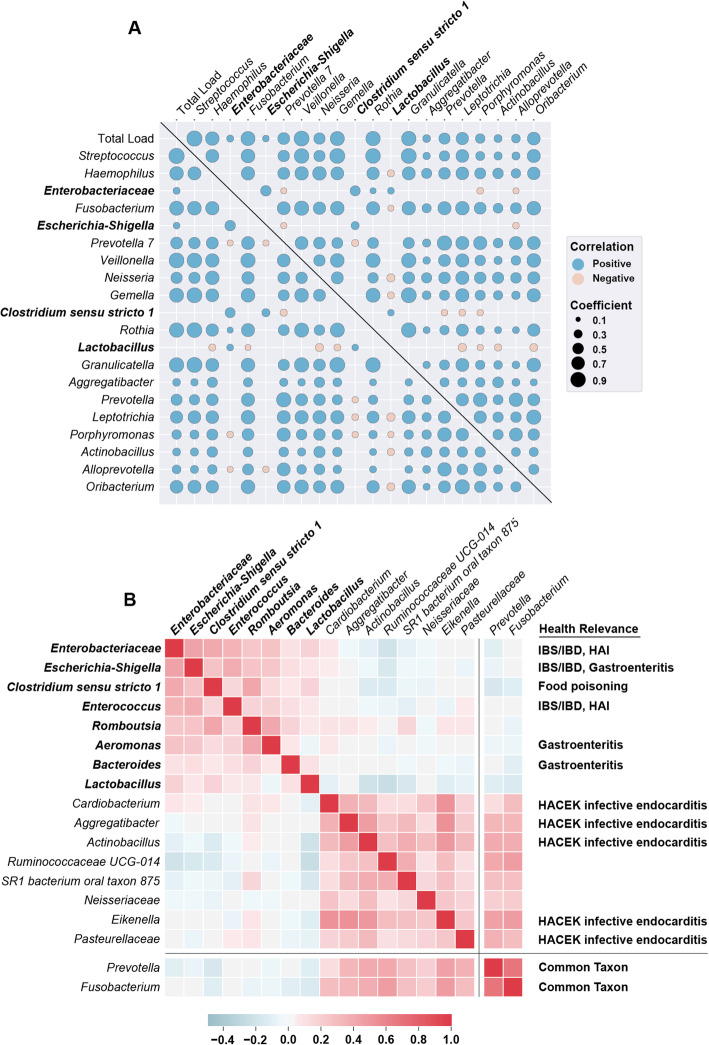
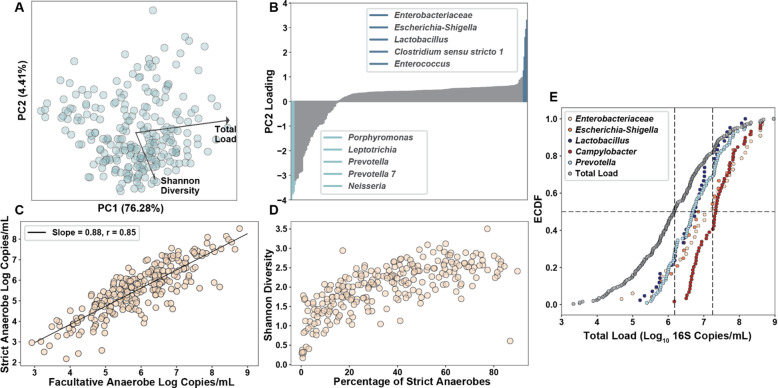
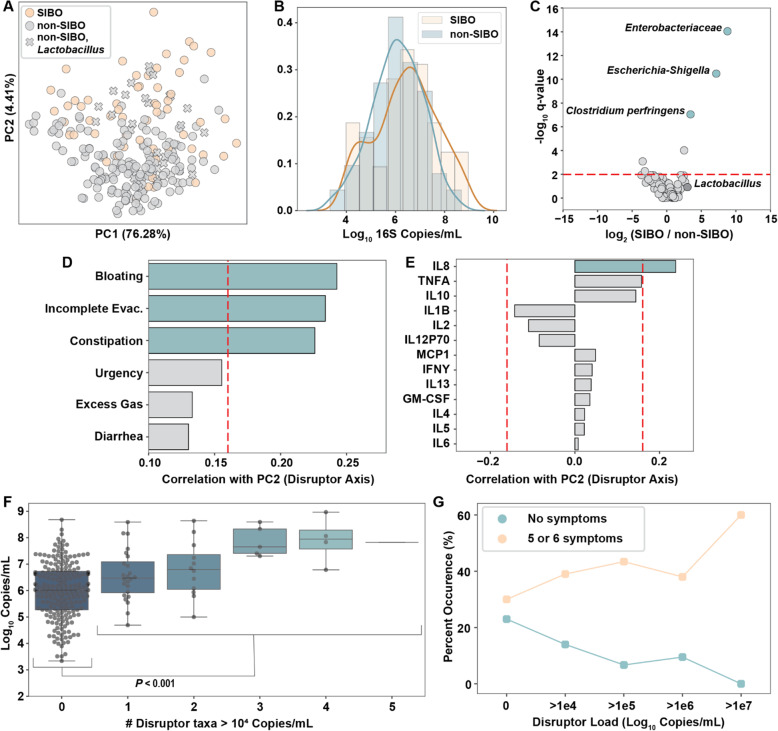
Results: Using quantitative sequencing, we evaluated total and taxon-specific absolute microbial loads from 250 duodenal-aspirate samples and 21 paired duodenum-saliva samples from participants in the REIMAGINE study. Log-transformed total microbial loads spanned 5 logs and were normally distributed. Paired saliva-duodenum samples suggested potential transmission of oral microbes to the duodenum, including organisms from the HACEK group. Several taxa, including Klebsiella, Escherichia, Enterococcus, and Clostridium, seemed to displace strict anaerobes common in the duodenum, so we refer to these taxa as disruptors. Disruptor taxa were enriched in samples with high total microbial loads and in individuals with small intestinal bacterial overgrowth (SIBO). Absolute loads of disruptors were associated with more severe GI symptoms, highlighting the value of absolute taxon quantification when studying small-intestine health and function.
Conclusion: This study provides the largest dataset of the absolute abundance of microbiota from the human duodenum to date. The results reveal a clear relationship between the oral microbiota and the duodenal microbiota and suggest an association between the absolute abundance of disruptor taxa, SIBO, and the prevalence of severe GI symptoms. Video Abstract.
Keywords: Bloating; Constipation; Duodenum; Enterobacteriaceae; HACEK; Human small intestinal microbiome; IBS; Lactobacillus; SIBO; Saliva.
© 2021. The Author(s).
Conflict of interest statement
The quantitative sequencing technology described in this publication is the subject of a patent application filed by Caltech. R.F.I. receives patent royalties from Bio-Rad related to droplet digital PCR.
Figures
References
-
- Yasuda K, Oh K, Ren B, Tickle TL, Franzosa EA, Wachtman LM, Miller AD, Westmoreland SV, Mansfield KG, Vallender EJ, Miller GM, Rowlett JK, Gevers D, Huttenhower C, Morgan XC. Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microbe. 2015;17(3):385–391. doi: 10.1016/j.chom.2015.01.015. - DOI - PMC - PubMed
-
- Leite GGS, Weitsman S, Parodi G, Celly S, Sedighi R, Sanchez M, Morales W, Villanueva-Millan MJ, Barlow GM, Mathur R, Lo SK, Jamil LH, Paski S, Rezaie A, Pimentel M. Mapping the segmental microbiomes in the human small bowel in comparison with stool: a REIMAGINE study. Dig Dis Sci. 2020;65(9):2595–2604. doi: 10.1007/s10620-020-06173-x. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
