

Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes
- PMID: 34725325
- PMCID: PMC8560912
- DOI: 10.1038/s41467-021-26502-6
Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes
Abstract
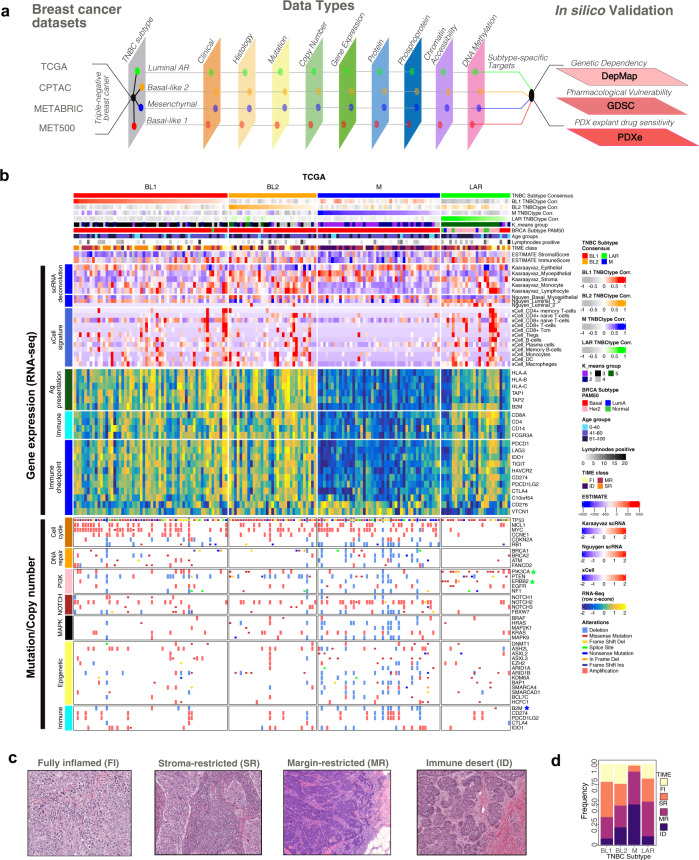
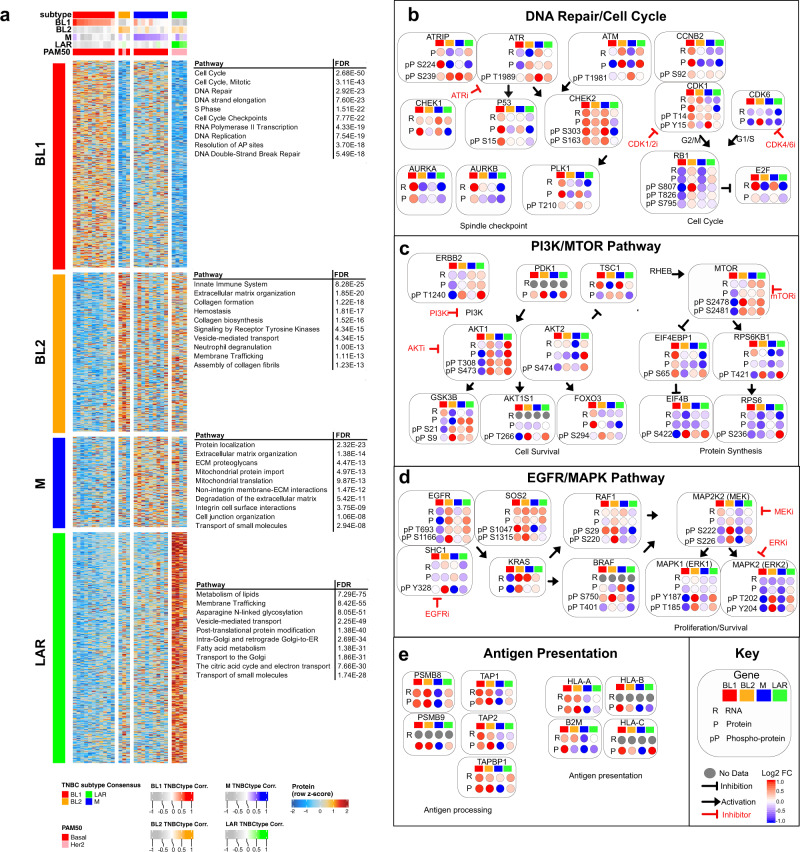
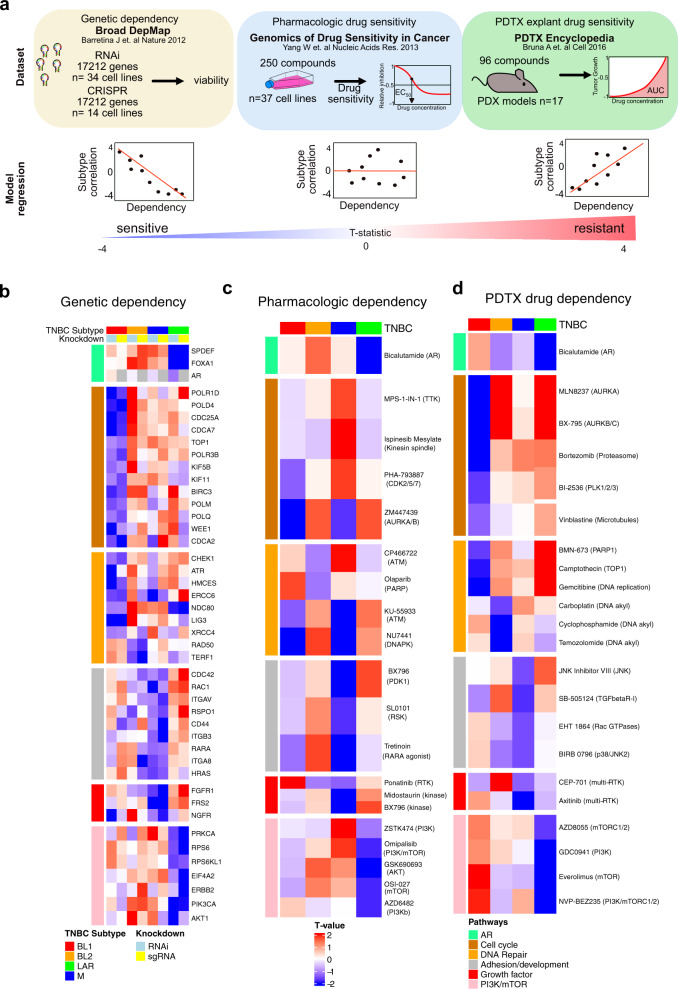
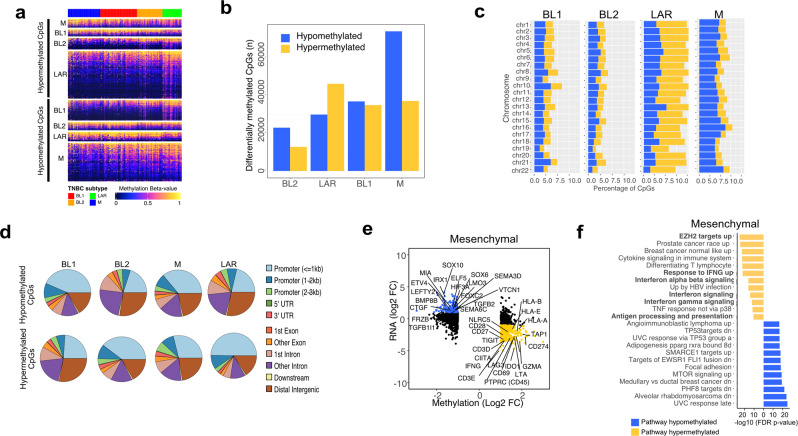
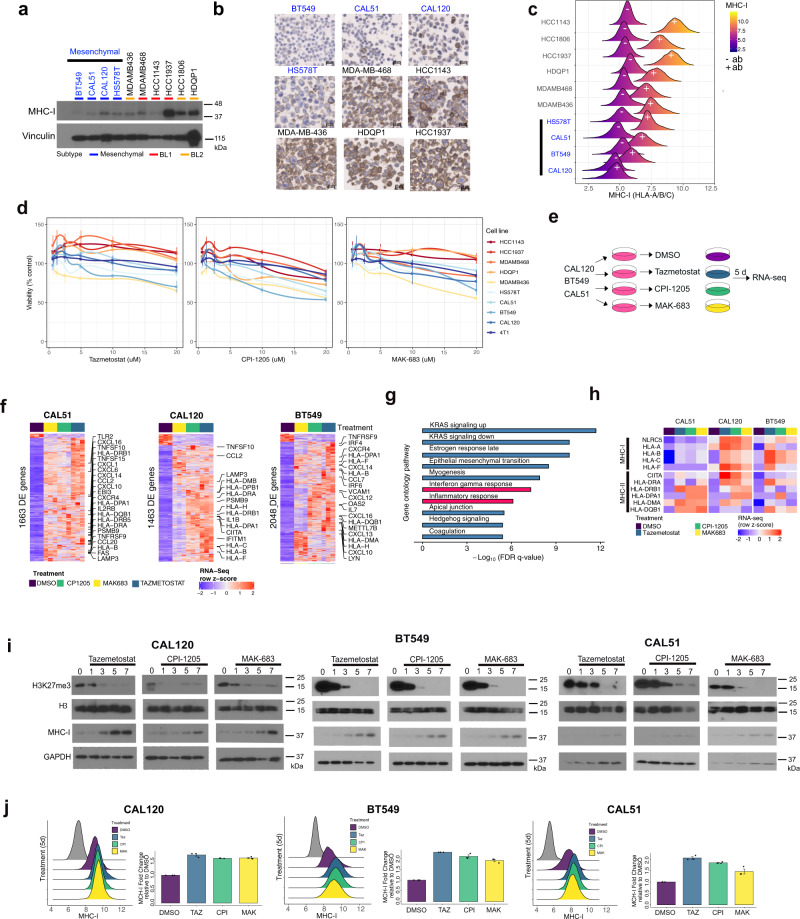
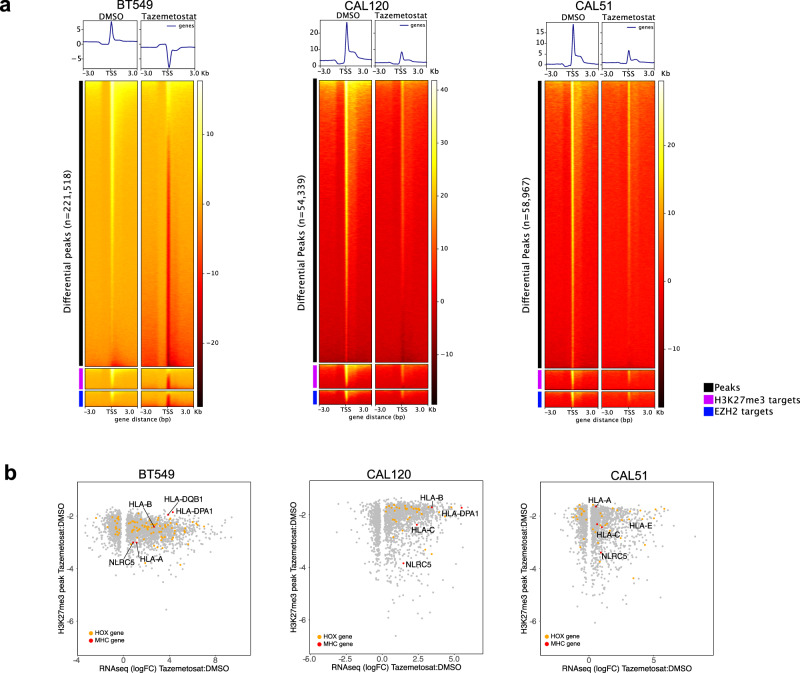
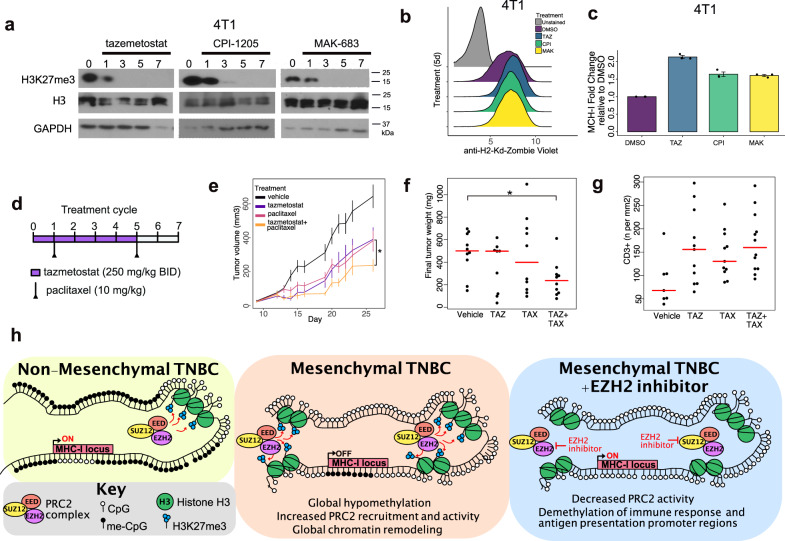
Triple-negative breast cancer (TNBC) is a collection of biologically diverse cancers characterized by distinct transcriptional patterns, biology, and immune composition. TNBCs subtypes include two basal-like (BL1, BL2), a mesenchymal (M) and a luminal androgen receptor (LAR) subtype. Through a comprehensive analysis of mutation, copy number, transcriptomic, epigenetic, proteomic, and phospho-proteomic patterns we describe the genomic landscape of TNBC subtypes. Mesenchymal subtype tumors display high mutation loads, genomic instability, absence of immune cells, low PD-L1 expression, decreased global DNA methylation, and transcriptional repression of antigen presentation genes. We demonstrate that major histocompatibility complex I (MHC-I) is transcriptionally suppressed by H3K27me3 modifications by the polycomb repressor complex 2 (PRC2). Pharmacological inhibition of PRC2 subunits EZH2 or EED restores MHC-I expression and enhances chemotherapy efficacy in murine tumor models, providing a rationale for using PRC2 inhibitors in PD-L1 negative mesenchymal tumors. Subtype-specific differences in immune cell composition and differential genetic/pharmacological vulnerabilities suggest additional treatment strategies for TNBC.
© 2021. The Author(s).
Conflict of interest statement
X.C., B.D.L. and J.A.P. are inventors (PCT/US2012/065724) of intellectual property (TNBCtype) licensed by Insight Genetics Inc. J.M.B. receives research support from Genentech/Roche, Bristol Myers Squibb, and Incyte Corporation, has received consulting/expert witness fees from Novartis, and is an inventor on provisional patents regarding immunotherapy targets and biomarkers in cancer. The remaining authors declare no competing interests.
Figures
References
-
- Schmid P, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:44–59. doi: 10.1016/S1470-2045(19)30689-8. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
