

Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface
- PMID: 34725363
- PMCID: PMC8560802
- DOI: 10.1038/s41467-021-26614-z
Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface
Abstract
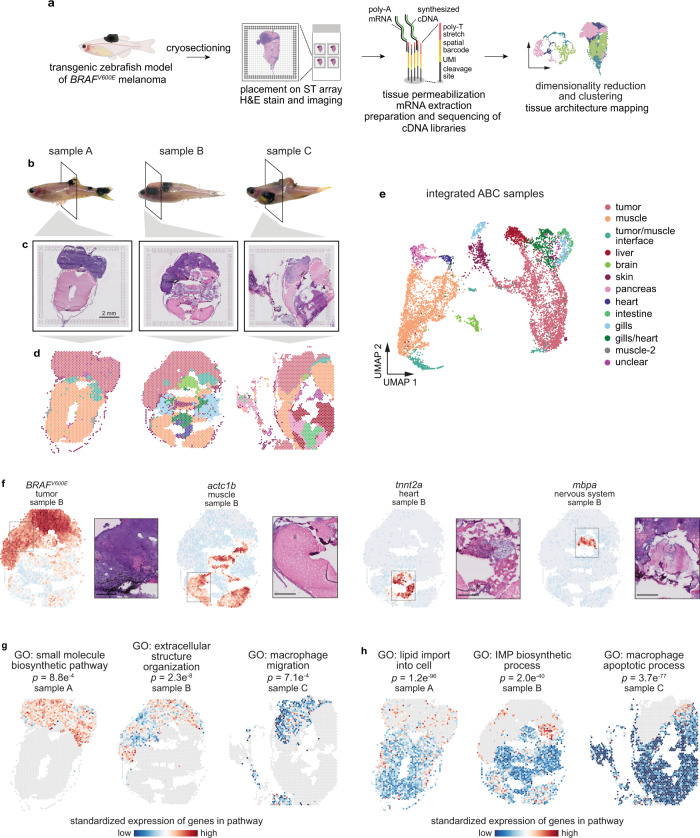
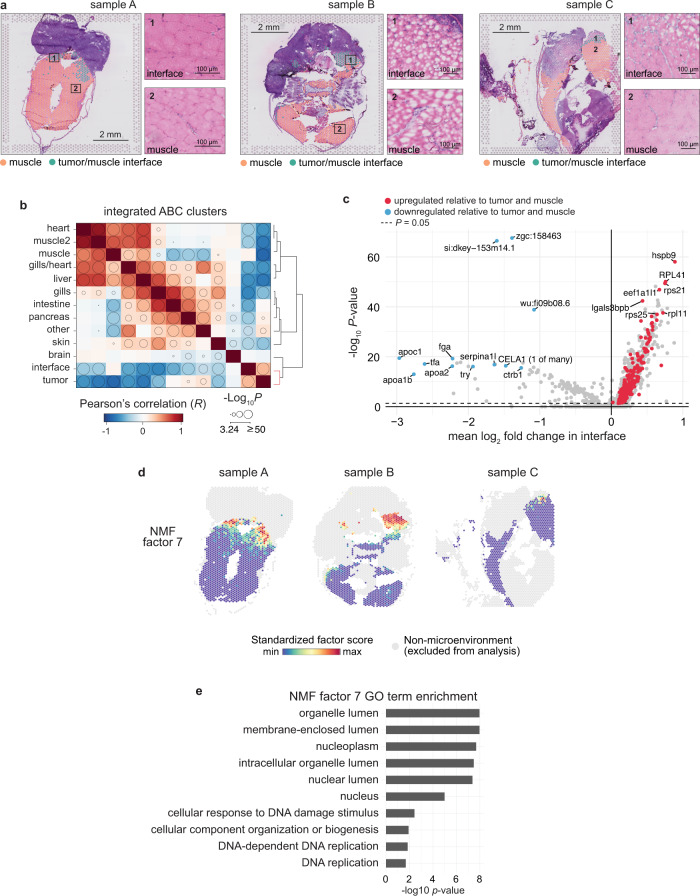
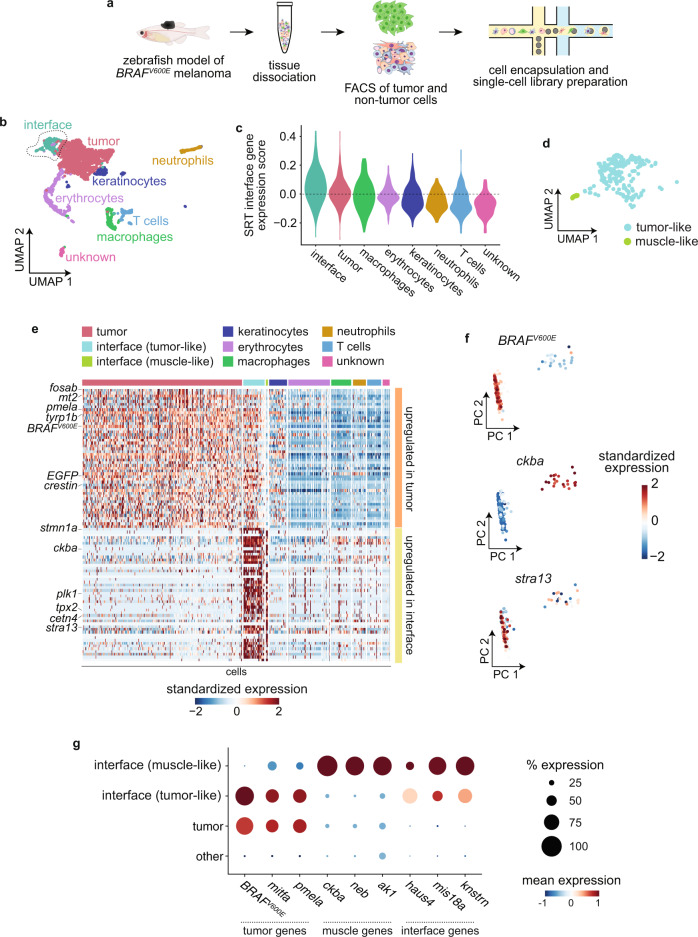
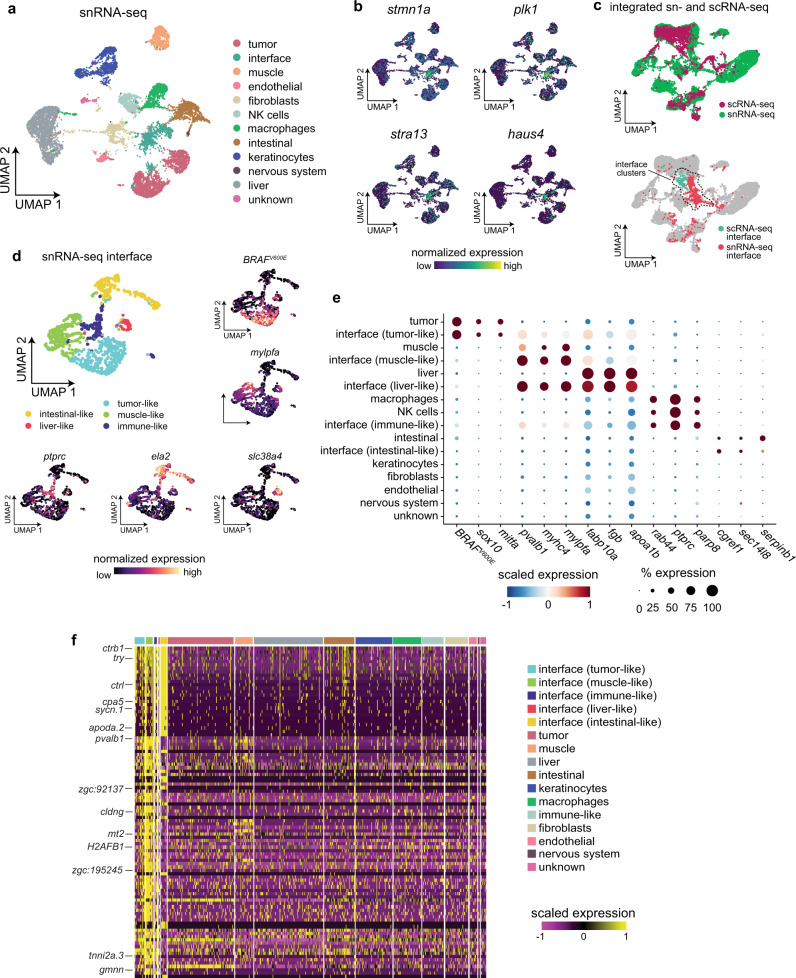
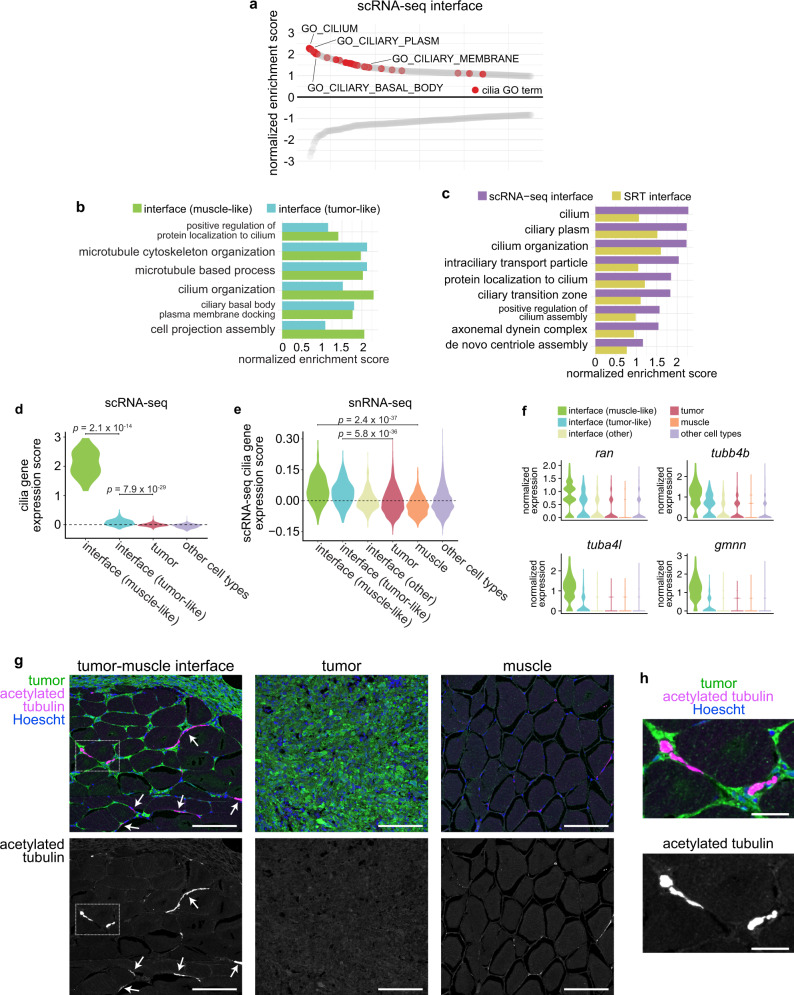
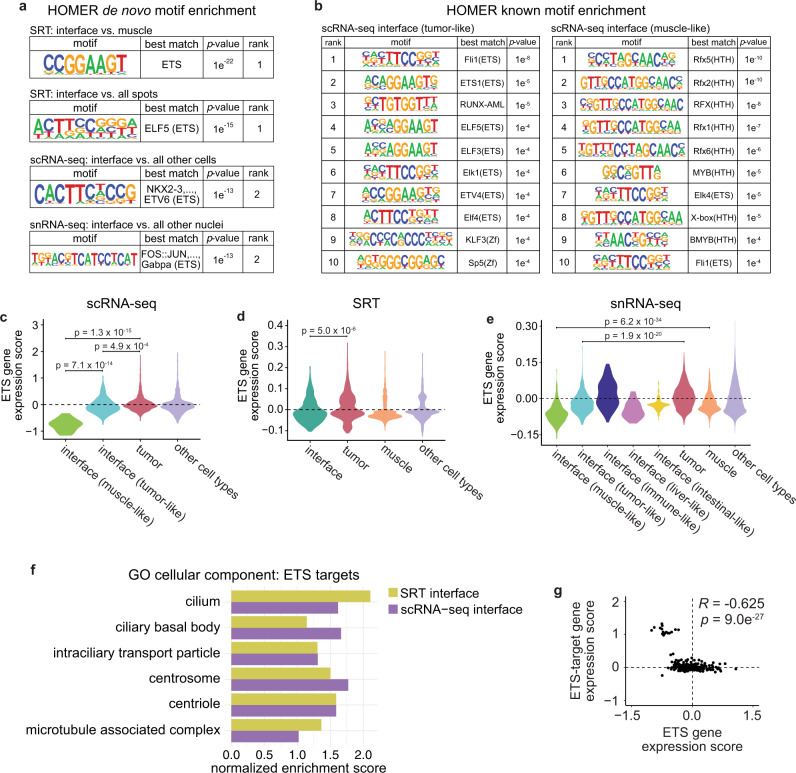
During tumor progression, cancer cells come into contact with various non-tumor cell types, but it is unclear how tumors adapt to these new environments. Here, we integrate spatially resolved transcriptomics, single-cell RNA-seq, and single-nucleus RNA-seq to characterize tumor-microenvironment interactions at the tumor boundary. Using a zebrafish model of melanoma, we identify a distinct "interface" cell state where the tumor contacts neighboring tissues. This interface is composed of specialized tumor and microenvironment cells that upregulate a common set of cilia genes, and cilia proteins are enriched only where the tumor contacts the microenvironment. Cilia gene expression is regulated by ETS-family transcription factors, which normally act to suppress cilia genes outside of the interface. A cilia-enriched interface is conserved in human patient samples, suggesting it is a conserved feature of human melanoma. Our results demonstrate the power of spatially resolved transcriptomics in uncovering mechanisms that allow tumors to adapt to new environments.
© 2021. The Author(s).
Conflict of interest statement
R.M.W. is a paid consultant to N-of-One Therapeutics, a subsidiary of Qiagen. R.M.W. is on the scientific advisory board of Consano, but receives no income for this. R.M.W. receives royalty payments for the use of the
Figures
References
-
- Zhang M, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 2018;8:1006–1025. doi: 10.1158/2159-8290.CD-17-1371. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
